Brugada syndrome
| Brugada syndrome | |
 | |
|---|---|
| ECG findings of Brugada Syndrome | |
| ICD-10 | I42.8 |
| ICD-9 | 746.89 |
| OMIM | 601144 |
| DiseasesDB | 31999 |
| eMedicine | med/3736 |
| MeSH | D053840 |
|
WikiDoc Resources for Brugada syndrome |
|
Articles |
|---|
|
Most recent articles on Brugada syndrome Most cited articles on Brugada syndrome |
|
Media |
|
Powerpoint slides on Brugada syndrome |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Brugada syndrome at Clinical Trials.gov Trial results on Brugada syndrome Clinical Trials on Brugada syndrome at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Brugada syndrome NICE Guidance on Brugada syndrome
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Brugada syndrome Discussion groups on Brugada syndrome Patient Handouts on Brugada syndrome Directions to Hospitals Treating Brugada syndrome Risk calculators and risk factors for Brugada syndrome
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Brugada syndrome |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Associate Editor-In-Chief: Cafer Zorkun, M.D., Ph.D. [2]
Please Join in Editing This Page and Apply to be an Editor-In-Chief for this topic: There can be one or more than one Editor-In-Chief. You may also apply to be an Associate Editor-In-Chief of one of the subtopics below. Please mail us [3] to indicate your interest in serving either as an Editor-In-Chief of the entire topic or as an Associate Editor-In-Chief for a subtopic. Please be sure to attach your CV and or biographical sketch.
Overview
The Brugada syndrome is a genetic disease that is characterized by abnormal electrocardiogram (ECG) findings and an increased risk of sudden cardiac death in young adults, and occasionally in children and infants. It is also known as Sudden Unexpected Death Syndrome[1] (SUDS), and is the most common cause of sudden death in young men without known underlying cardiac disease in Thailand and Laos[2].
Although the ECG findings of Brugada syndrome were first reported[3] among survivors of cardiac arrest in 1989, it was only in 1992 that the Brugada brothers[4] recognized it as a distinct clinical entity, causing sudden death by causing ventricular fibrillation (a lethal arrhythmia) in the heart.
Differential Diagnosis
- Abnormalities that can lead to ST-segment elevation in the right precordial leads
- Acute myocardial ischemia or infarction
- Acute myocarditis
- Acute pulmonary thromboemboli
- Arrhythmogenic right ventricular dysplasia / cardiomyopathy (ARVD/C)
- Cocaine intoxication
- Dissecting aortic aneurysm
- Duchenne muscular dystrophy
- Friedreich ataxia
- Heterocyclic antidepressant overdose
- Hypercalcemia
- Hyperkalemia
- Hypothermia, causing Osborn wave in ECGs and sometimes resembling Brugada syndrome
- Left ventricular hypertrophy
- Mediastinal tumor compressing the right ventricular outflow tract (RVOT)
- Right or left bundle-branch block
- Right ventricular infarction
- Right ventricular ischemia
- Thiamine deficiency
- Various central and autonomic nervous system abnormalities
- Early repolarization syndrome
- Other normal variants (particularly in males)
Epidemiology
The average age at the time of initial diagnosis or sudden death is 40 ± 22 years, with the youngest patient diagnosed at 2 days of age and the oldest at 84 years. The prevalence of the Brugada syndrome is estimated at 1–5 per 10,000 inhabitants worldwide. The frequency is lower in western countries and higher (≥5 per 10,000) in Southeast Asia.
Genetics and pathophysiology
Approximately 20% of the cases of Brugada syndrome have been shown to be associated with mutation(s) in the gene that encodes for the sodium ion channel in the cell membranes of the muscle cells of the heart (the myocytes). The gene, named SCN5A, is located on the short arm of the third chromosome (3p21). Loss-of-function mutations in this gene lead to a loss of the action potential dome of some epicardial areas of the right ventricle. This results in transmural and epicardial dispersion of repolarization. The transmural dispersion underlies ST-segment elevation and the development of a vulnerable window across the ventricular wall, whereas the epicardial dispersion of repolarization facilitates the development of phase 2 reentry, which generates a phase 2 reentrant extrasystole that captures the vulnerable window to precipitate ventricular tachycardia and/or fibrillation that often results in sudden cardiac death. At present time however, all the reported patients died because of the disease and submitted to detailed necropsy study, have shown a structural right ventricular pathology underlying the syndrome.
| Type | OMIM | Mutation | Notes |
| B1 | 601144 | alpha subunit of the sodium channel (SCN5A) | Current through this channel is commonly referred to as INa. Gain of this channel leads to an unopposed Ito current (KCND2) |
| B2 | 611778 | GPD1L, Glycerol-3-phosphate dehydrogenase like peptide | |
| B3 | 114205 | CACNA1C | Alpha subunit of cardiac L-type calcium channel.[5] |
| B4 | 600003 | CACNB2 | Beta-2 subunit of the voltage dependent L-type calcium channel.[5] |
| B5 | 604433 | KCNE3 which coassembles with KCND3 | Beta subunit to KCND3. Modulates the Ito potassium outward current[6] |
| B6 | 600235 | SCN1B | Beta-1 subunit of the sodium channel SCN5A[7] |
Over 160 mutations in the SCN5A gene have been discovered to date, each having varying mechanisms and effects on function, thereby explaining the varying degrees of penetration and expression of this disorder. [8]
An example of one of the mechanisms in which a loss of function of the sodium channel occurs is a mutation in the gene that disrupts the sodium channel's ability to bind properly to ankyrin-G, an important protein mediating interaction between ion channels and cytoskeletal elements. Very recently a mutation in a second gene, Glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) has been shown to result in Brugada Syndrome in a large multigenerational family (London, 2006). This gene acts as an ion channel modulator in the heart, although the exact mechanism is not yet understood.
Recently Antzelevitch has identified mutations in the L-type calcium channel subunits (CACNA1C (A39V and G490R) and CACNB2 (S481L)) leading to ST elevation and a relatively short QT interval (below 360 msec).[9]
This condition is inherited in an autosomal dominant pattern and is more common in males. In addition it has a higher prevalence in most Asian populations.[10] [11] [12] [13] [14] [15] [16]
Electrocardiography
In some cases, the disease can be detected by observing characteristic patterns on an electrocardiogram, which may be present all the time, or might be elicited by the administration of particular drugs (e.g., Class IC antiarrythmic drugs that blocks sodium channels and causing appearance of ECG abnormalities - ajmaline, flecainide) or resurface spontaneously due to as yet unclarified triggers. The pattern seen on the ECG is persistent ST elevations in the electrocardiographic leadsV1-V3 with a right bundle branch block (RBBB) appearance with or without the terminal S waves in the lateral leads that are associated with a typical RBBB. A prolongation of the PR interval (a conduction disturbance in the heart) is also frequently seen.The electrocardiogram can fluctuate over time, depending on the autonomic balance and the administration of antiarrhythmic drugs. Adrenergic stimulation decreases the ST segment elevation, while vagal stimulation worsens it. (There is a case report of a patient who died while shaving, presumed due to the vagal stimulation of the carotid sinus massage!) The administration of class Ia, Ic and III drugs increases the ST segment elevation, and also fever. Exercise decreases ST segment elevation in some patients but increases it in others (after exercise when the body temperature has risen). The changes in heart rate induced by atrial pacing are accompanied by changes in the degree of ST segment elevation. When the heart rate decreases, the ST segment elevation increases and when the heart rate increases the ST segment elevation decreases. However, the contrary can also be observed.
Characteristics
- Characterized by a coved-type ST-segment elevation in the right precordial leads
- The Brugada ECG is often concealed, but can be unmasked or modulated by a number of drugs and pathophysiological states including sodium channel blockers, a febrile state, vagotonic agents, tricyclic antidepressants, as well as cocaine and Propranolol intoxication.
Genetics
- SCN5A is a gene that encodes the alpha sodium unit of the cardiac sodium channel. Mutations in SCN5A account for about 15-30% of Brugada syndrome cases. A negative genetic test for SCN5A does not exclude that SCN5A is causing the clinical syndrome because the genetic tests do not evaluate for mutations in promotors, cryptic splicing mutations, or gross rearrangements in the protein product.
- Glycerol-3-phosphate dehydrogenase (GPD1L) is associated with progressive conduction disease and low sensitivity to procainamide resulting from decreased Isodium current. It has a relatively good prognosis.
- CACNA1C (alpha subunit of L-type cardiac calcium channel) and CACNB2b (beta subunit of L-type cardiac calcium channel) is associated with a shortened QT interval and a combinatin Brugada/Short QT interval syndrome.
Brugada EKG
- Type 1 ST segment elevation is diagnostic of Brugada syndrome and is characterized by a coved ST-segment elevation ≥2 mm (0.2 mV) followed by a negative T wave.
- Type 2 ST-segment elevation has a saddleback appearance with a high take-off ST-segment elevation of ≥2 mm followed by a trough displaying ≥1 mm ST elevation followed by either a positive or biphasic T wave.
- Type 3 ST-segment elevation has either a saddleback or coved appearance with an ST-segment elevation of <1 mm.
-
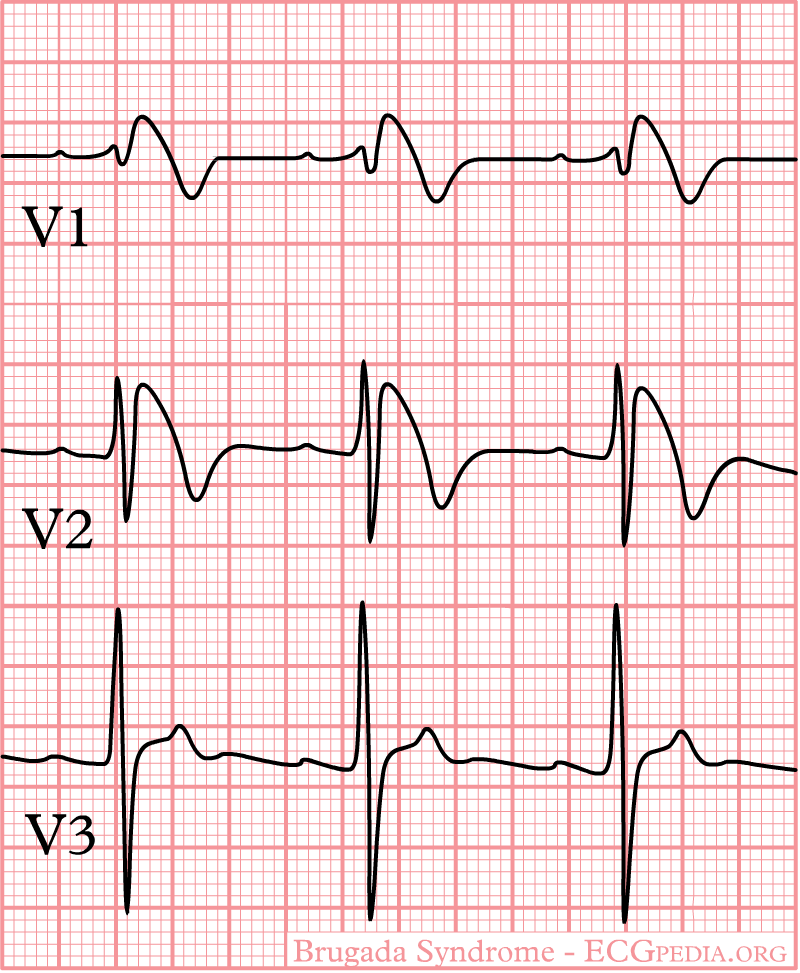
General characteristics
-

EKG of a Patient with Brugada Syndrome
-
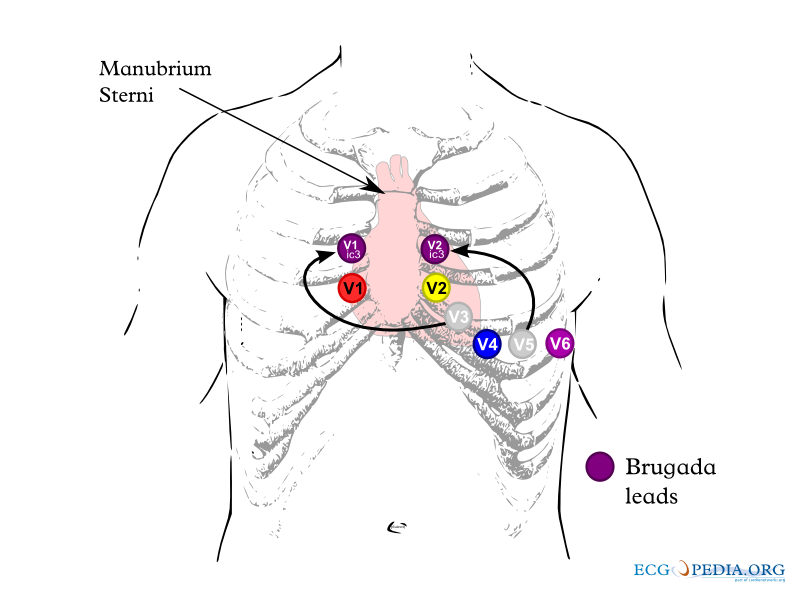
Lead placements


-
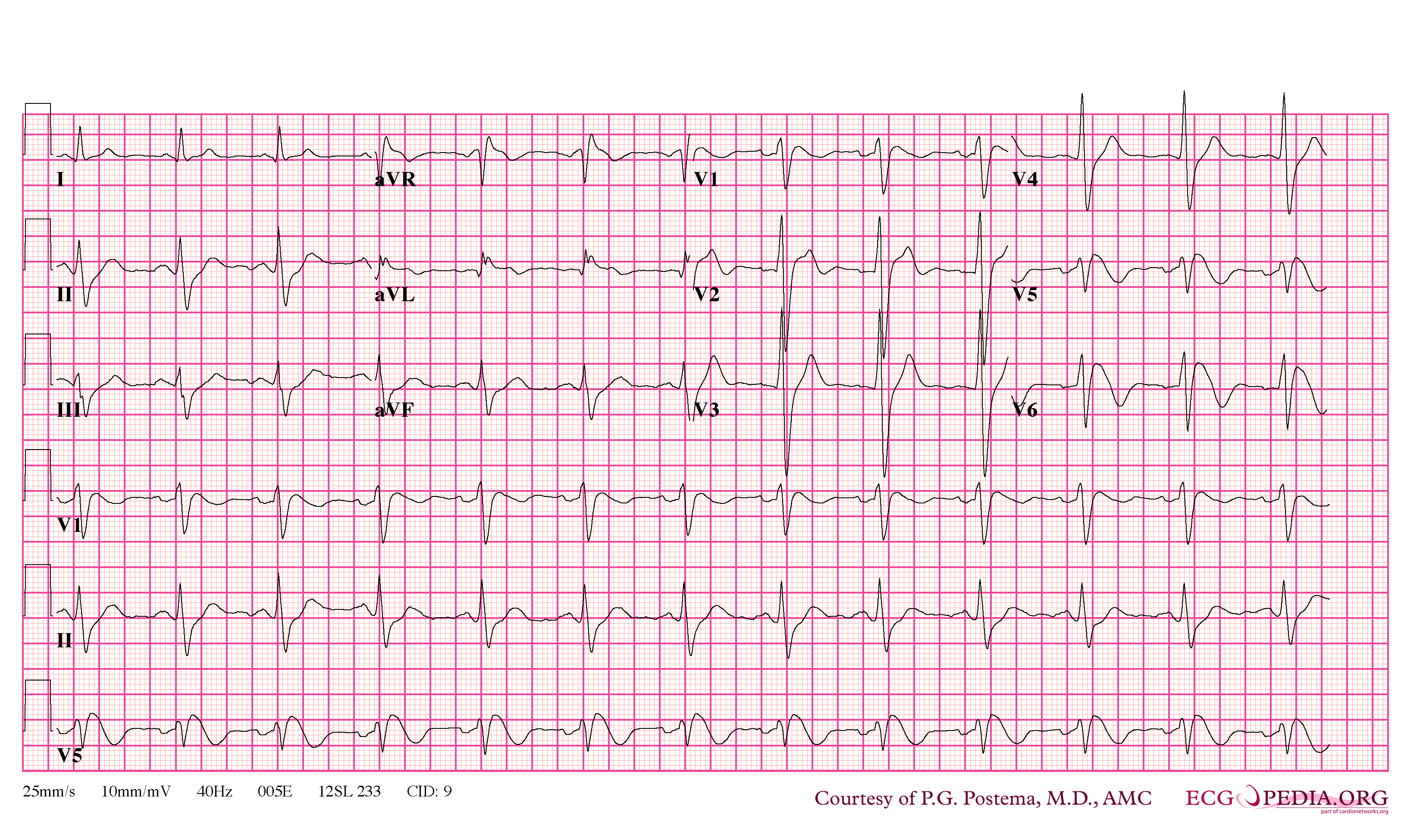
Brugada Type 1
-
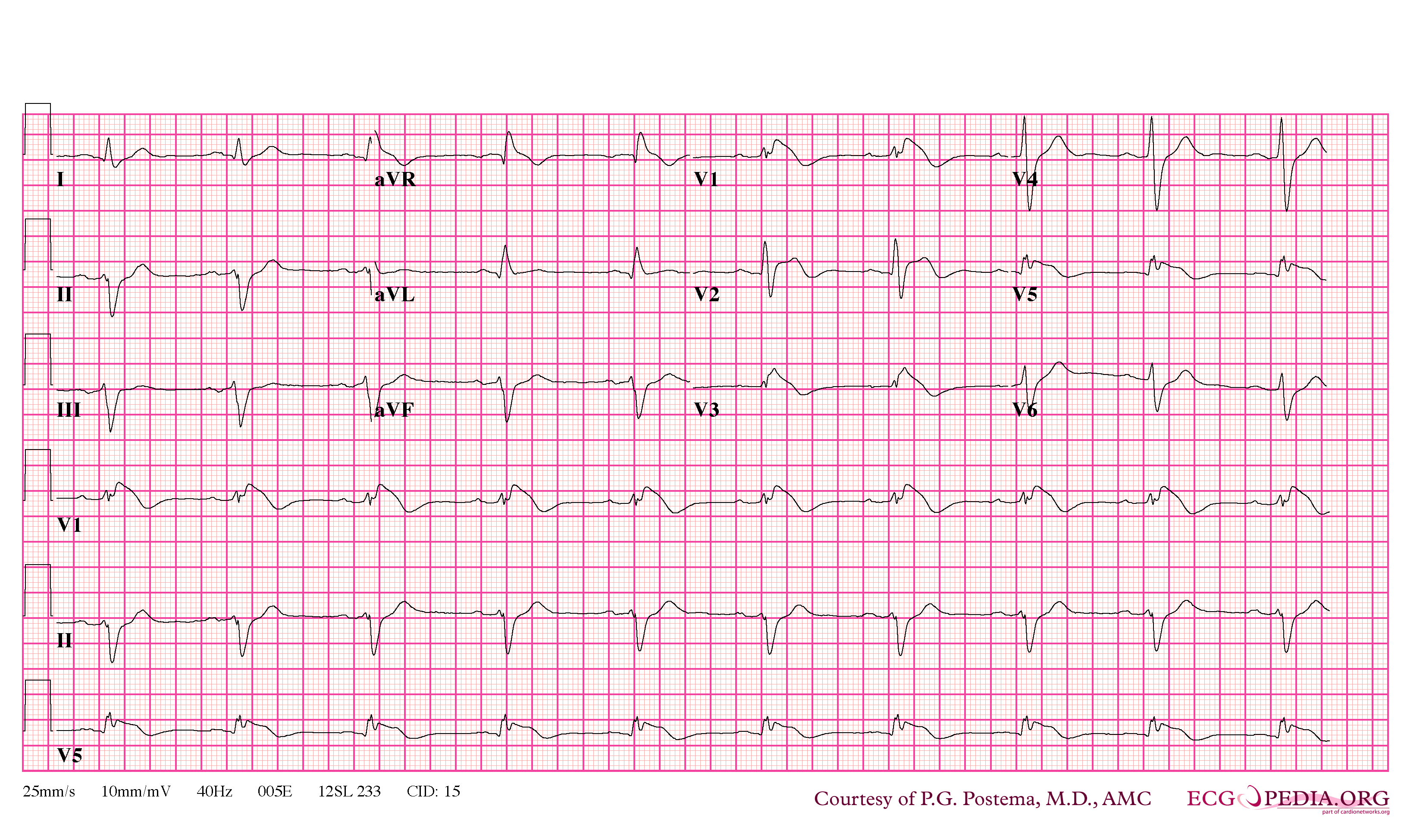
Brugada Type 1
-
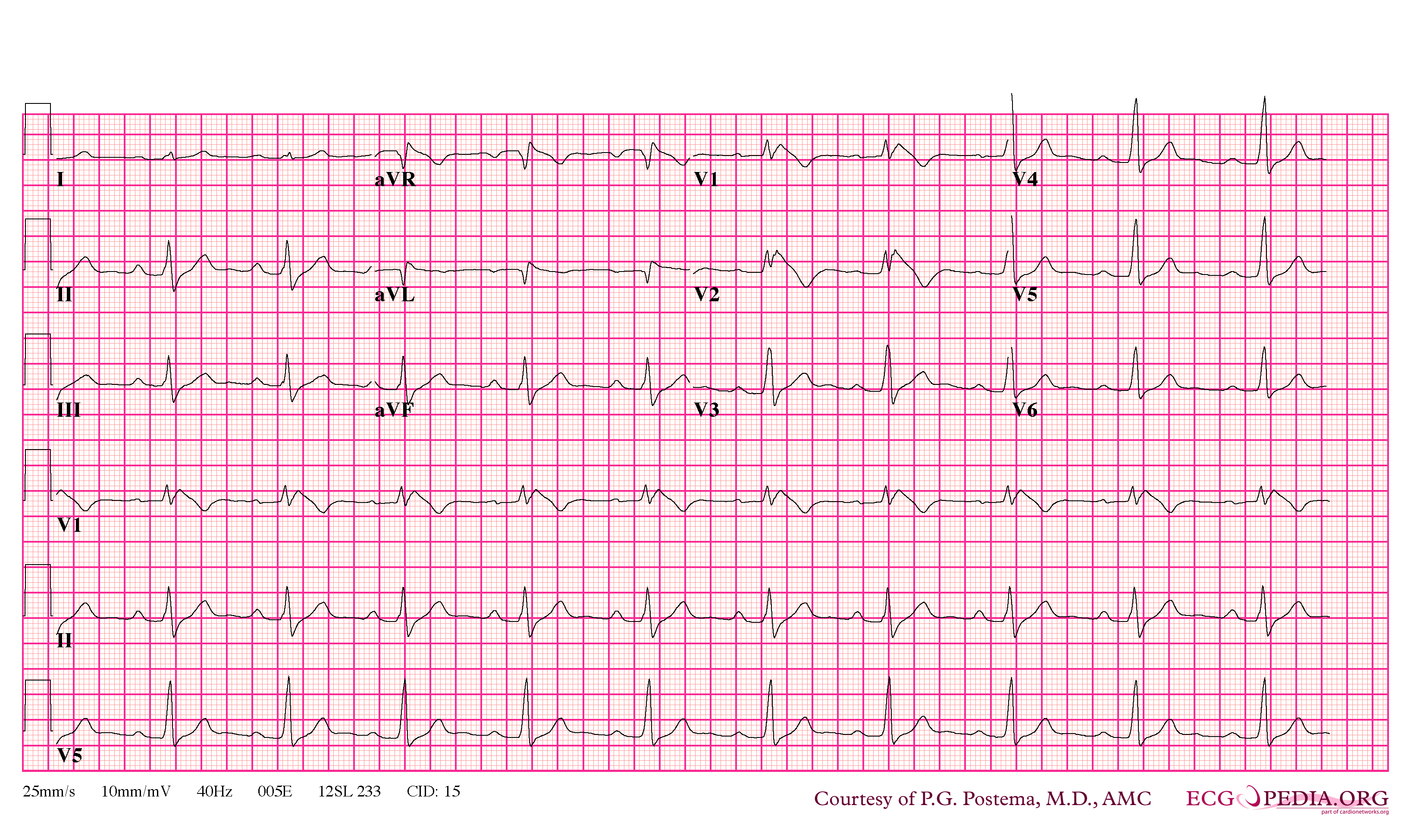
Brugada Type 1



-
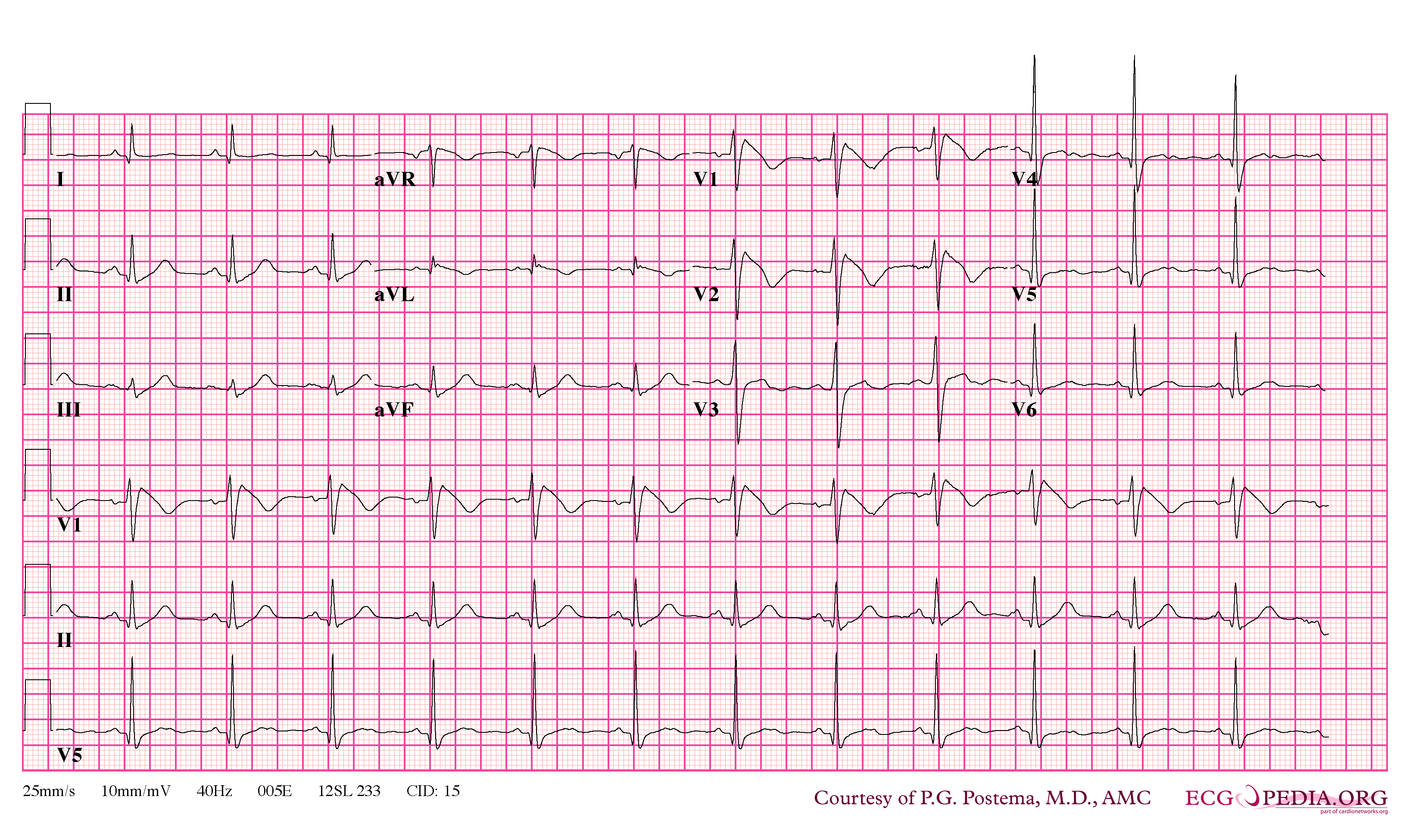
Brugada Type 1
-
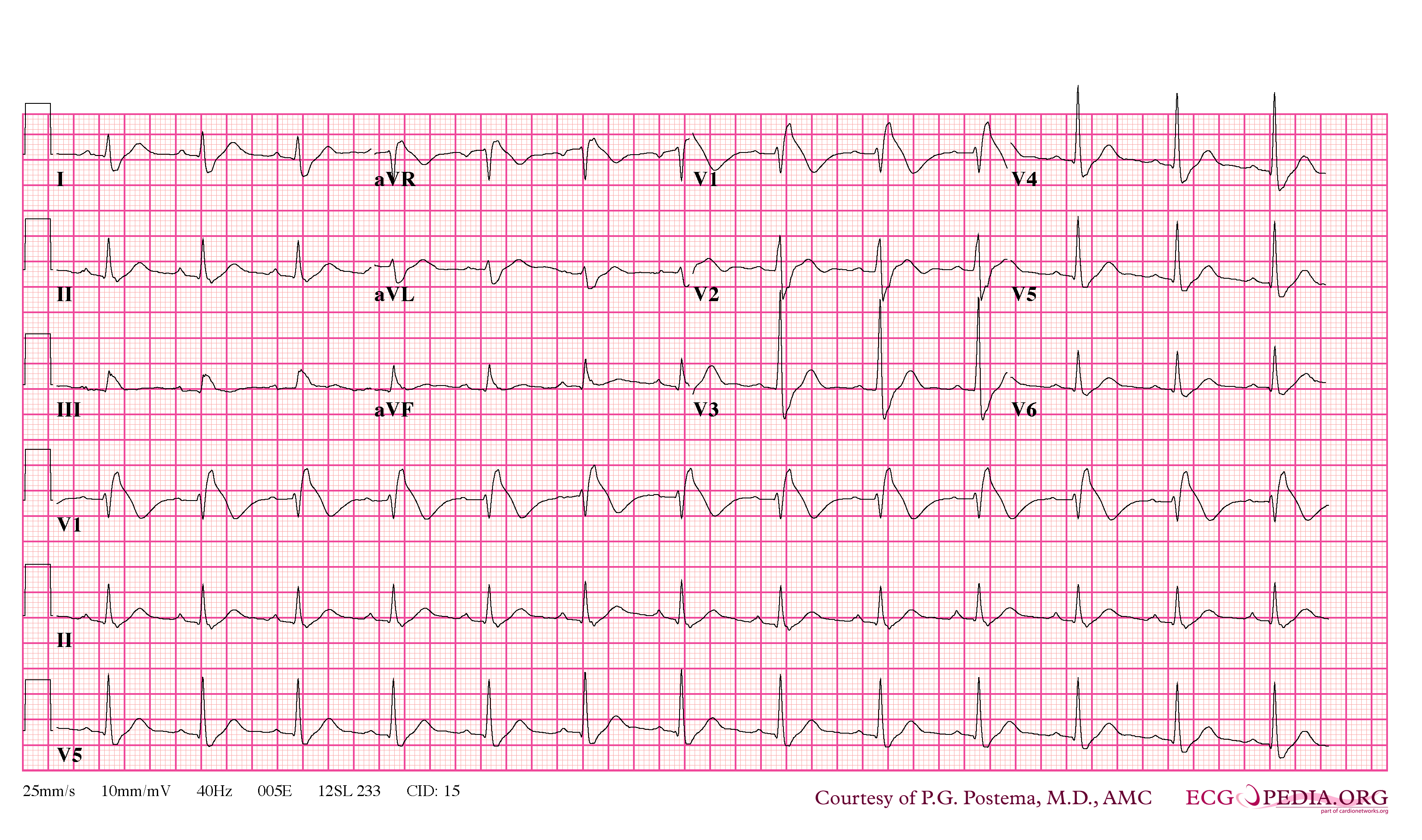
Brugada Type 1
-

Brugada Type 1



-
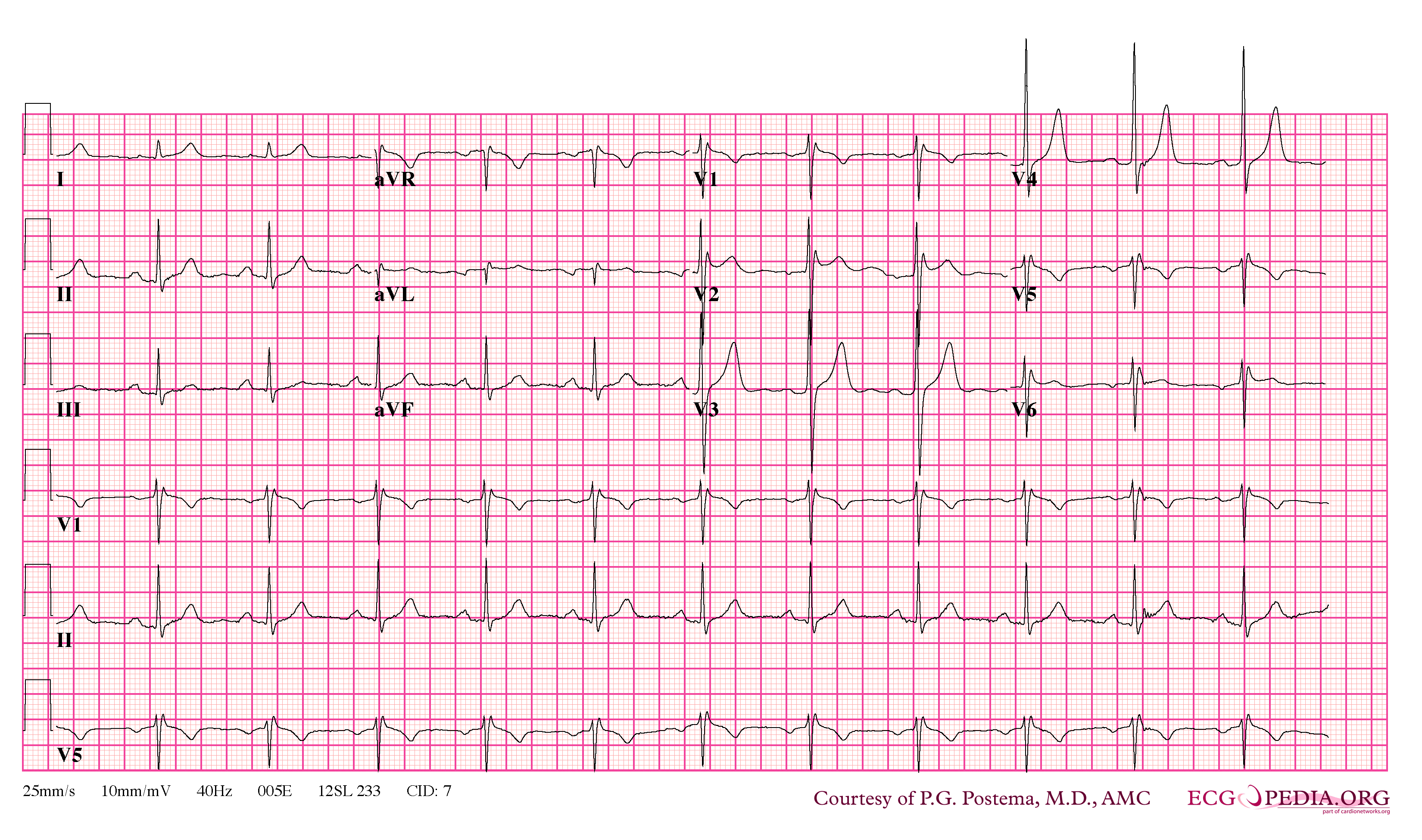
Brugada Type 2
-
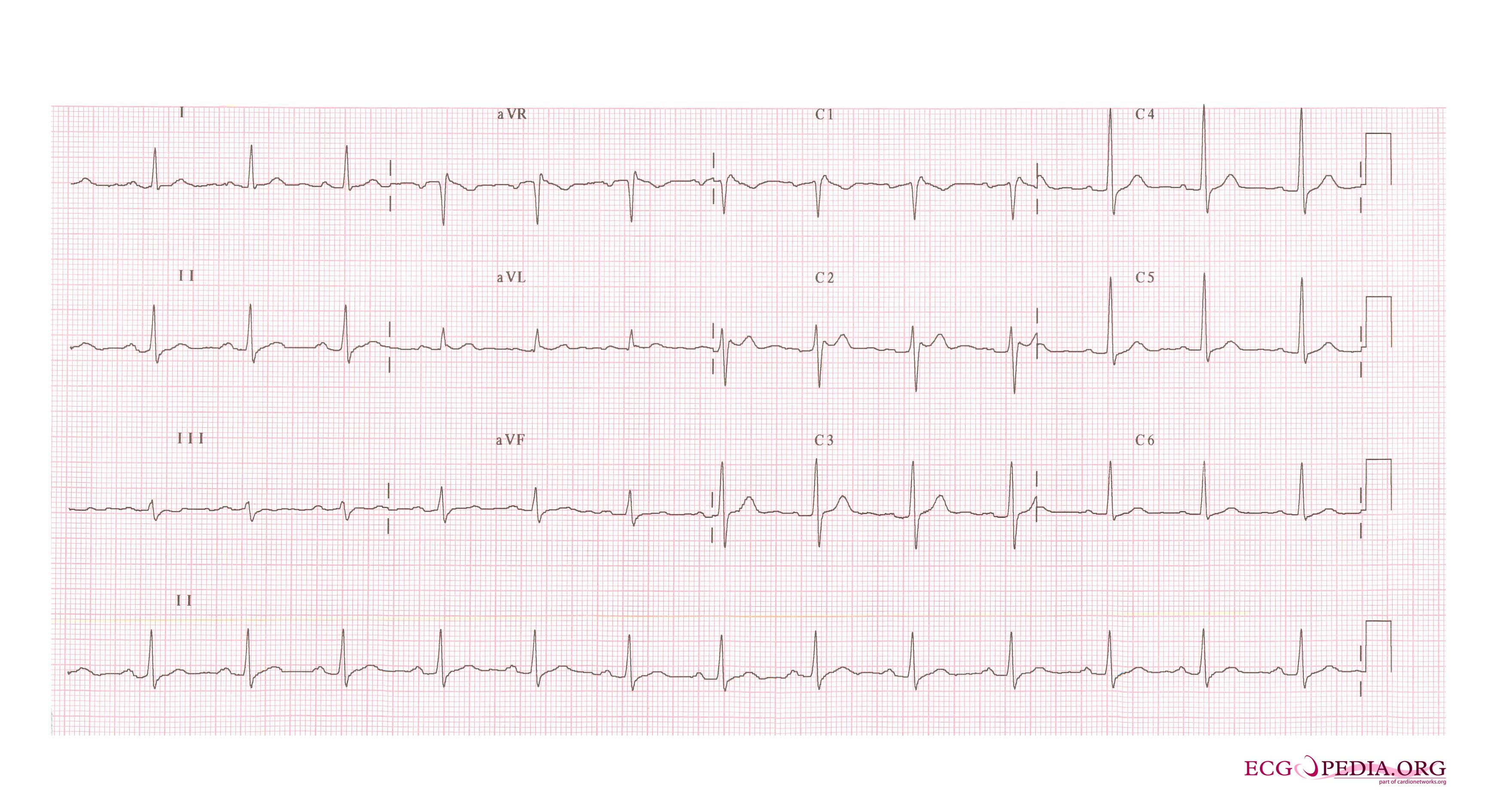
Brugada Type 2


{kind=link}
Diagnosis
- Diagnosed when a Type 1 ST-segment elevation is observed in more than one right precordial lead (V1-V3), in the presence or absence of sodium channel blocking agent, and in conjunction with one or more of the following:
- Family history of SCD (<45 years old)
- Documented VF
- Polymorphic ventricular tachycardia
- Coved-type ECGs in family members
- Inducibility of VT with programmed electrical stimulation (PES)
- Syncope
- Nocturnal agonal respiration
- Diagnosis is also considered positive when a Type 2 (saddleback pattern) or Type 3 ST-segment elevation is observed in more than one right precordial lead under baseline conditions and can be converted to the diagnostic Type 1 pattern occurs upon exposure to sodium channel blocker.
Sodium Challenge
- Drugs that can be used
- Ajmaline 1 mg/kg/5 min IV
- Flecainide 2 mg/kg/10 min IV or 400 mg PO
- Procainamide 10 mg/kg/10 min IV
- Pilsicainide 1 mg/kg/10 min IV
- The sodium challenge should be terminated when
- Diagnostic Type 1 ST-segment elevation or Brugada ECG, develops
- ST segment in Type 2 increases by ≥2 mm
- Premature ventricular beats or other arrhythmias develop
- QRS widens to ≥130% of baseline
Arrhythmias
- Polymorphic VT resembling a rapid Torsade de Pointes (TdP)
- Monomorphic VT is observed infrequently
- VT/VF often terminates spontaneously in patients with the Brugada syndrome which may explain why patients wake up at night after episodes of agonal respiration caused by the arrhythmia.
Risk Statification
- Patients with syncope and an abnormal Type 1 ECG are at higher risk
- Asymptomatic patients at risk can be identified
- Presence of spontaneous Type 1 ST-segment elevation
- Characteristics of the S wave
- Presence of late potentials
- Inducibility of VT/VF using PES.
Treatment
The cause of death in Brugada syndrome is ventricular fibrillation.The episodes of syncope (fainting) and sudden death (aborted or not) are caused by fast polymorphic ventricular tachycardias or ventricular fibrillation. These arrhythmias appear with no warning. While there is no exact treatment modality that reliably and totally prevents ventricular fibrillation from occurring in this syndrome, treatment lies in termination of this lethal arrhythmia before it causes death. This is done via implantation of an implantable cardioverter-defibrillator (ICD), which continuously monitors the heart rhythm and will defibrillate an individual if ventricular fibrillation is noted. Some recently performed studies had evaluated the role of quinidine, a Class Ia antiarrythmic drug, for decreasing VF episodes occurring in this syndrome. Quinidine was found to decrease number of VF episodes and correcting spontaneous ECG changes, possibly via inhibiting Ito channels.[17] Those with risk factors for coronary artery disease may require an angiogram before ICD implantation.
- Aborted sudden death are at high risk for recurrence and should receive an ICD
- VT storm has been successfully treated with Isoproterenol. The mechanism is thought to be augmenting the cardiac L type channel.
- Asymptomatic patients require risk stratification and clinical judegement to help guide therapy
- Quinidine (class IA sodium channel blocker) blocks the Ito current and is proven to suppress spontaneous VF
- Cilostazol (phosphodiesterase III inhibitor that increases inward L type calcium channel current and reported to suppress spontaneous VF
- Bepridil suppress spontaneous VF probably through blocking Ito current
- Medical therapy alone with the above agents is currently not evaluated in randomized trials and should not be used as loan therapy.
See also
References
- ↑ Hong K, Berruezo-Sanchez A, Poungvarin N, Oliva A, Vatta M, Brugada J, Brugada P, Towbin JA, Dumaine R, Pinero-Galvez C, Antzelevitch C, Brugada R. Phenotypic characterization of a large European family with Brugada syndrome displaying a sudden unexpected death syndrome mutation in SCN5A. J Cardiovasc Electrophysiol. 2004 Jan;15(1):64-9. PMID 15028074
- ↑ Brugada J, Brugada P, Brugada R. The syndrome of right bundle branch block ST segment elevation in V1 to V3 and sudden death--the Brugada syndrome. Europace. 1999 Jul;1(3):156-66. PMID 11225790
- ↑ Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, Daliento L, Dalla Volta S. Ventricular fibrillation without apparent heart disease: description of six cases. Am Heart J 1989 Dec;118(6):1203-9 PMID 2589161
- ↑ Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992 Nov 15;20(6):1391-6. PMID 1309182
- ↑ 5.0 5.1 Antzelevitch C, Pollevick GD, Cordeiro JM; et al. (2007). "Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death". Circulation. 115 (4): 442–229. doi:10.1161/CIRCULATIONAHA.106.668392. PMID 17224476.
- ↑ Delpon E, Cordeiro JM, Núñez L; et al. (2008). "Functional Effects of KCNE3 Mutation and Its Role in the Development of Brugada Syndrome". Circulation Arrhythmia and Electrophysiology. 1 (3): 209–18. doi:10.1161/CIRCEP.107.748103. PMID 19122847.
- ↑ Watanabe H, Koopmann TT, Le Scouarnec S; et al. (2008). "Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans". J. Clin. Invest. 118 (6): 2260–8. doi:10.1172/JCI33891. PMC 2373423. PMID 18464934. Unknown parameter
|month=ignored (help) - ↑ Napolitano C, Priori SG (2006). "Brugada syndrome". Orphanet journal of rare diseases. 1: 35. doi:10.1186/1750-1172-1-35. PMID 16972995.
- ↑ Antzelevitch C (2007). "Genetic basis of Brugada syndrome". Heart rhythm : the official journal of the Heart Rhythm Society. 4 (6): 756–7. doi:10.1016/j.hrthm.2007.03.015. PMID 17556198.
- ↑ Brugada Syndrome. Charles Antzelevitch, PH.D. PACE 2006; 29:1130–1159
- ↑ Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J Am Coll Cardiol 1992; 20:1391–1396.
- ↑ Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, Perez Riera AR. Brugada syndrome. A decade of progress. Circ Res 2002; 91:1114–1119.
- ↑ Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Eur Heart J 2002; 23:1648–1654.
- ↑ Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation 2002; 106:2514–2519.
- ↑ Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome. Report of the second consensus conference. Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- ↑ Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome:Report of the second consensus conference. Heart Rhythm 2005; 2:429–440.
- ↑ Belhassen B, Glick A, Viskin S (2004). "Efficacy of quinidine in high-risk patients with Brugada syndrome". Circulation. 110 (13): 1731–7. doi:10.1161/01.CIR.0000143159.30585.90. PMID 15381640.
External links
- GeneReviews: Brugada syndrome
- Algado et al: http://www.medspain.com/ant/n13_jun00/Brugada.htm
- Behr: http://www.c-r-y.org.uk/long_qt_syndrome.htm
- The Ramon Brugada Senior Foundation
- http://digilander.libero.it/martini_syndrome/
Template:SIB
de:Brugada-Syndrom
it:Sindrome di Brugada
he:תסמונת ברוגדה
fi:Brugadan oireyhtymä