Pulmonary hypertension
For patient information click here
| Pulmonary hypertension | |
| ICD-10 | I27.0, I27.2 |
|---|---|
| ICD-9 | 416 |
| DiseasesDB | 10998 |
| MeSH | D006976 |
Editors-in-Chief: Richard Channick, M.D.; C. Michael Gibson, M.S., M.D. [1] Phone:617-632-7753
Assistant Editor-in-Chief: Ann Slater, R.N.
Overview
Pulmonary hypertension (PH) is an increase in blood pressure in the pulmonary artery or lung vasculature, leading to shortness of breath, dizziness, fainting, and other symptoms, all of which are exacerbated by exertion. Depending on the cause, pulmonary hypertension can be a severe disease with a markedly decreased exercise tolerance and right-sided heart failure. It was first identified by Dr Ernst von Romberg in 1891.[1] It can be one of five different types, arterial, venous, hypoxic, thromboembolic, or miscellaneous.
Although the terms primary pulmonary hypertension (meaning of unknown cause) and secondary pulmonary hypertension (meaning due to another medical condition) still persist in materials disseminated to patients and the general public, these terms have largely been abandoned in the medical literature. This change has occurred because the older dichotomous classification did not reflect pathophysiology or outcome. It led to erroneous therapeutic decisions, i.e. treat "primary" pulmonary hypertension only. This in turn led to therapeutic nihilism for many patients labeled "secondary" pulmonary hypertension, and could have contributed to their deaths. The term "primary pulmonary hypertension" has now been replaced with "idiopathic pulmonary arterial hypertension". The terms "primary" and "secondary" pulmonary hypertension should not be used any longer. Further details are in the Classification section below.
Signs and symptoms
A history usually reveals gradual onset of shortness of breath, fatigue, non-productive cough, angina pectoris, fainting or syncope, peripheral edema (swelling of the limbs, especially around the ankles and feet), and rarely hemoptysis (coughing up blood). Pulmonary arterial hypertension (PAH) typically does not present with orthopnea or paroxysmal nocturnal dyspnea, while pulmonary venous hypertension typically does.
In order to establish the cause, the physician will generally conduct a thorough medical history. A detailed family history is taken to determine whether the disease might be familial. A history of exposure to cocaine, methamphetamine, alcohol leading to cirrhosis, and smoking leading to emphysema are considered significant. Physical examination is performed to look for typical signs of pulmonary hypertension including a loud P2 (pulmonic valve closure sound), (para)sternal heave, jugular venous distension, pedal edema, ascites, hepatojugular reflux, clubbing etc. Physical examination evidence of tricuspid insufficiency is also sought and, if present, is consistent with the presence of long-standing pulmonary hypertension.
Differential Diagnosis of the Causes of Pulmonary Hypertension
- Alveolar hypoventilation
- Atrial septal defect
- Bronchiectasis
- COPD
- Collagen vascular diseases
- Cystic fibrosis
- Infection
- Interstitial lung disease
- Portal hypertension
- Pulmonary embolism
- Pulmonary venous obstruction
- Sarcoidosis
- Scleroderma
- Sickle cell disease
- Sleep apnea
Causes
The most common cause of pulmonary hypertension is left heart failure leading to pulmonary venous hypertension. This may be due to systolic or diastolic malfunction of the left ventricle or due to valvular dysfunction such as mitral regurgitation or mitral stenosis. It usually manifests as pulmonary edema. Because the malfunctioning heart does not pump efficiently, blood fails to leave the pulmonary circulation in a timely manner, leading to abnormally high pressure in the pulmonary vein.[2] The increased pressure in the pulmonary vein can be carried through to the pulmonary artery.[2]
Common causes of pulmonary arterial hypertension (PAH) include HIV, scleroderma and other autoimmune disorders, cirrhosis and portal hypertension, sickle cell disease,[3] congenital heart disease, and others. Use of weight loss pills such as Fen-Phen, Aminorex, fenfluramine (Pondimin), and phentermine led to the development of PAH in the past.[4] Other causes include sarcoidosis, histiocytosis X, and fibrosing mediastinitis.
Pulmonary embolism also leads to pulmonary hypertension, acutely as well as chronically. When none of these causes can be found, the disease is termed idiopathic pulmonary arterial hypertension (IPAH). There appears to be a link between IPAH and thyroid diseases,[5], but this is not regarded as causative.
Lung diseases that lower oxygen in the blood (hypoxia) are well known causes of pulmonary hypertension, including COPD, interstitial lung disease, Pickwickian syndrome or obesity-hypoventilation syndrome, and possibly sleep apnea. Human herpesvirus 8, also associated with Kaposi's sarcoma, has been demonstrated in patients with PAH, suggesting that this virus may play a role in its development.[6] Recent studies have been unable to find an association between human herpesvirus 8 and idiopathic pulmonary arterial hypertension.
When a family history exists, the disease is termed familial pulmonary arterial hypertension (FPAH). IPAH and FPAH are now considered to be genetic disorders linked to mutations in the BMPR2 gene, which encodes a receptor for bone morphogenetic proteins,[7] as well as the 5-HT(2B) gene, which codes for a serotonin receptor.[8]
There seems to be an association of idiopathic PAH (not only PAH caused by heart malformations) and Trisomy 21.
Pathogenesis
Whatever the initial cause, pulmonary hypertension involves the tightening of blood vessels connected to and within the lungs. This makes it harder for the heart to pump blood through the lungs, much as it is harder to make water flow through a narrow pipe as opposed to a wide one. Over time, the affected blood vessels become both stiffer and thicker, further increasing the blood pressure within the lungs and impairing blood flow. In addition, the increased workload of the heart causes thickening and enlargement of the right ventricle, making the heart less able to pump blood through the lungs, causing right heart failure. As the blood flowing through the lungs decreases, the left side of the heart receives less blood. This blood may also carry less oxygen than normal. Therefore it becomes harder and harder for the left side of the heart to pump to supply sufficient oxygen to the rest of the body, especially during physical activity.
Diagnosis
Because pulmonary hypertension can be of 5 major types, a series of tests must be performed to distinguish pulmonary arterial hypertension from venous, hypoxic, thomboembolic, or miscellaneous varieties.
A physical examination is performed to look for typical signs of pulmonary hypertension. These include altered heart sounds, such as a widely split S2 or second heart sound, a loud P2 or pulmonic valve closure sound (part of the second heart sound), (para)sternal heave, possible S3 or third heart sound, and pulmonary regurgitation. Other signs include jugular venous distension (enlargement of the jugular veins), peripheral edema (swelling of the ankles and feet), ascites (abdominal swelling due to the accumulation of fluid), hepatojugular reflux, and clubbing.
Further procedures are required to confirm the presence of pulmonary hypertension and exclude other possible diagnoses. These generally include pulmonary function tests, blood tests, electrocardiography (ECG), arterial blood gas measurements, X-rays of the chest (followed by high-resolution CT scanning if interstitial lung disease is suspected), and ventilation-perfusion or V/Q scanning to exclude chronic thromboembolic pulmonary hypertension. Biopsy of the lung is usually not indicated unless the pulmonary hypertension is thought to be due to an underlying interstitial lung disease. But lung biopsies are fraught with risks of bleeding due to the high intrapulmonary blood pressure. Clinical improvement is often measured by a "six-minute walk test", i.e. the distance a patient can walk in six minutes. Stability and improvement in this measurement correlate with better survival.
Although pulmonary arterial pressure can be estimated on the basis of echocardiography, pressure sampling with a Swan-Ganz catheter provides the most definite measurement. PAOP and PVR can not be measured directly with echocardiography. Therefore diagnosis of PAH requires a cardiac catheterization. A Swan-Ganz catheter can also measure the cardiac output, which is far more important in measuring disease severity than the pulmonary arterial pressure.
Normal pulmonary arterial pressure in a person living at sea level has a mean value of 12–16 mm Hg (1600–2100 Pa). Definite pulmonary hypertension is present when mean pressures at rest exceed 25 mm Hg (3300 Pa). If mean pulmonary artery pressure rises above 30 mm Hg (4000 Pa) with exercise, that is also considered pulmonary hypertension.
Diagnosis of PAH requires the presence of pulmonary hypertension with two other conditions. Pulmonary artery occlusion pressure (PAOP or PCWP) must be less than 15 mm Hg (2000 Pa) and pulmonary vascular resistance (PVR) must be greater than 3 Wood units (240 dyn•s•cm-5 or 2.4 mN•s•cm-5).
Multi Sliced CT
Images shown below are courtesy of RadsWiki and copylefted
-
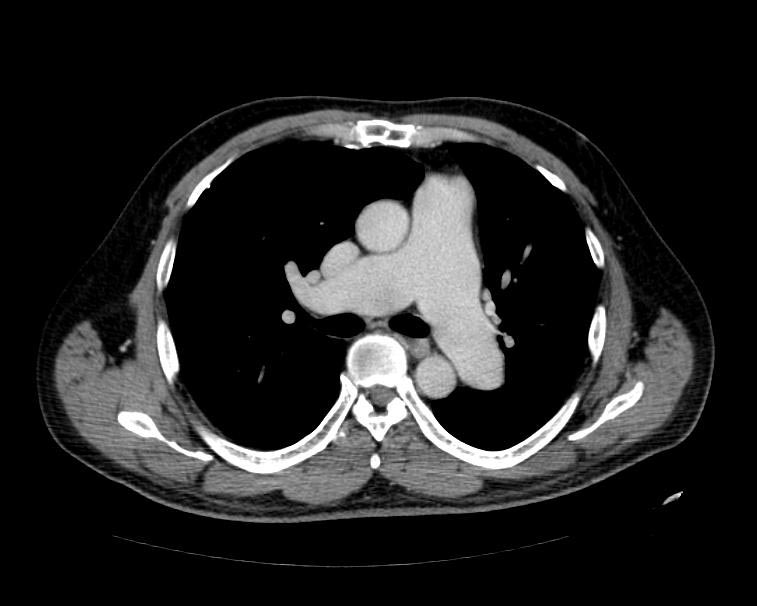 MSCT: Pulmonary hypertension. Note increase in diameter of pulmonary artery.
MSCT: Pulmonary hypertension. Note increase in diameter of pulmonary artery. -
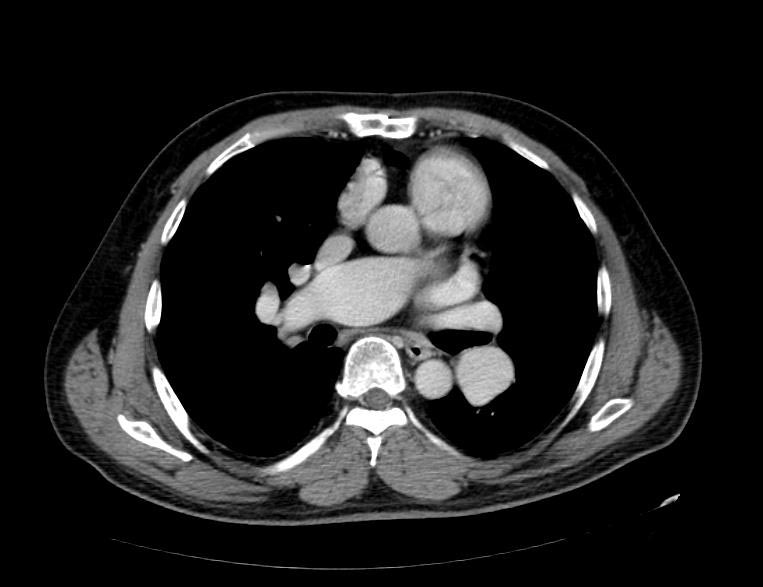 MSCT: Pulmonary hypertension. Note increase in diameter of pulmonary artery.
MSCT: Pulmonary hypertension. Note increase in diameter of pulmonary artery.


Classification
Current classification
In 2003, the 3rd World Symposium on Pulmonary Arterial Hypertension was convened in Venice to modify the classification based on the new understanding of disease mechanisms. The revised system developed by this group provides the current framework for understanding pulmonary hypertension.
The system includes several improvements over the former 1998 Evian Classification system. Risk factor descriptions were updated, and the classification of congenital systemic-to pulmonary shunts was revised. A new classification of genetic factors in PH was recommended, but not implemented because available data were judged to be inadequate.
The Venice 2003 Revised Classification system can be summarized as follows:[9]
- WHO Group I - Pulmonary arterial hypertension (PAH)
- WHO Group II - Pulmonary hypertension associated with left heart disease
- WHO Group III - Pulmonary hypertension associated with lung diseases and/or hypoxemia
- WHO Group IV - Pulmonary hypertension due to chronic thrombotic and/or embolic disease
- WHO Group V - Miscellaneous
Previous terminology
The terms primary and secondary pulmonary hypertension (PPH and SPH) were formerly used to classify the disease. This led to the assumption that only the primary disease should be treated, and the secondary variety should be ignored in favor of treating only the underlying illness. In fact all forms of pulmonary arterial hypertension are treatable. Unfortunately, this classification system still persists in the minds of many physicians, and probably leads to many patients with being denied treatment. This approach to pulmonary arterial hypertension may also contribute to underdiagnosis. It is estimated that there are about 100,000 patients with PAH in the US, but only 15-20,000 have been diagnosed. Many others have been misdiagnosed as COPD, asthma, or congestive heart failure.
The term primary pulmonary hypertension (PPH) has now been replaced with idiopathic pulmonary arterial hypertension (IPAH) in much of the medical literature. However, some physicians continue to use the older classification inappropriately.
Epidemiology
IPAH is a rare disease with an incidence of about 2-3 per million per year and a prevalence of about 15 per million. Women are almost three times as likely to present with IPAH than men.
Other forms of PAH are far more common. In scleroderma the incidence has been estimated to be 6 to 60% of all patients, in rheumatoid arthritis up to 21%, in systemic lupus erythematosus 4 to 14%, in portal hypertension between 2 to 5%, in HIV about 0.5%, and in sickle cell disease ranging from 20 to 40%.
Diet pills such as Fen-Phen produced an annual incidence of 25-50 per million per year.
Treatment
Treatment is determined by whether the PH is arterial, venous, hypoxic, thromboembolic, or miscellaneous. Since pulmonary venous hypertension is synonymous with congestive heart failure, the treatment is to optimize left ventricular function by the use of diuretics, beta blockers, ACE inhibitors, etc., or to repair/replace the mitral valve or aortic valve.
In PAH, lifestyle changes, digoxin, diuretics, oral anticoagulants, and oxygen therapy are considered conventional therapy, but have never been proven to be beneficial in a randomized, prospective manner.
High dose calcium channel blockers are useful in only 5% of IPAH patients who are vasoreactive by Swan-Ganz catheter. Unfortunately, calcium channel blockers have been largely misused, being prescribed to many patients with non-vasoreactive PAH, leading to excess morbidity and mortality.
Vasoactive substances
Three major pathways are involved in the abnormal proliferation and contraction of the smooth-muscle cells of the pulmonary artery in patients with pulmonary arterial hypertension. These pathways correspond to important therapeutic targets in this condition and play a role in determining which of three classes of drugs — endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and prostacyclin derivatives — will be used.
Prostaglandins
Prostacyclin (prostaglandin I2) is commonly considered the most effective treatment for PAH. Epoprostenol (synthetic prostacyclin, marketed as Flolan®) is given via continuous infusion that requires a semi-permanent central venous catheter. This delivery system can cause sepsis and thrombosis. Flolan® is unstable, and therefore has to be kept on ice during administration. Since it has a half-life of 3 to 5 minutes, the infusion has to be continuous (24/7), and interruption can be fatal. Other prostanoids have therefore been developed. Treprostinil (Remodulin®) can be given intravenously or subcutaneously, but the subcutaneous form can be very painful. Iloprost (Ilomedin®) is also used in Europe intravenously and has a longer half life. Iloprost (marketed as Ventavis®) is the only inhaled form of prostacyclin approved for use in the US and Europe. This form of administration has the advantage of selective deposition in the lungs with less systemic side effects.
Endothelin receptor antagonists
The dual (ETA and ETB) endothelin receptor antagonist bosentan (marketed as Tracleer®) was approved in 2001. Approved in June 2007, ambrisentan is marketed as Letairis® in U.S. by Gilead Sciences. Sitaxsentan has not been approved for marketing by the US FDA. A new trial is being planned to address FDA's concerns.
Phosphodiesterase type 5 inhibitors
Sildenafil, a selective inhibitor of cGMP specific phosphodiesterase type 5 (PDE5), was approved for the treatment of PAH in 2005. It is marketed for PAH as Revatio®. Tadalafil (currently marketed as Cialis® for erectile dysfunction) is currently is Phase III clinical trials.
Other agents
Vasoactive intestinal peptide by inhalation should enter clinical trials for PAH in 2007. PRX-08066 is a serotonin antagonist currently being developed for hypoxic pulmonary hypertension.
Surgical
Atrial septostomy is a surgical procedure that creates a communication between the right and left atria. It relieves pressure on the right side of the heart, but at the cost of lower oxygen levels in blood (hypoxia). It is best performed in experienced centers. Lung transplantation cures pulmonary arterial hypertension, but leaves the patient with the complications of transplantation, and a survival of about 5 years.
Pulmonary thromboendarterectomy (PTE) is a surgical procedure that is used for chronic thromboembolic pulmonary hypertension. It is the surgical removal of an organized thrombus (clot) along with the lining of the pulmonary artery; it is a large and very difficult procedure that is currently performed in a few select centers. Case series show remarkable success in most patients.
Treatment for hypoxic and miscellaneous varieties of pulmonary hypertension have not been established. However, studies of several agents are currently enrolling patients. Many physicians will treat these diseases with the same medications as for PAH, until better options become available.
Prognosis
The NIH IPAH registry from the 1980's showed an untreated median survival of 2-3 years from time of diagnosis, with the cause of death usually being right ventricular failure (cor pulmonale). Although this figure is widely quoted, it is probably irrelevant today. Outcomes have changed dramatically over the last two decades. This may be because of newer drug therapy, better overall care, and earlier diagnosis (lead time bias). A recent outcome study of those patients who had started treatment with bosentan (Tracleer®) showed that 86% patients were alive at 3 years. With multiple agents now available, combination therapy is increasingly used. Impact of these agents on survival is not known, since many of them have been developed only recently. It would not be unreasonable to expect median survival to extend past 10 years in the near future.
References
- ↑ Romberg E von. Über Sklerose der Lungenarterie. Dtsch Arch Klin Med 1891-1892;48:197-206.
- ↑ 2.0 2.1 Hayes, Gail Boyer. Pulmonary Hypertension Patient's Survival Guide 3rd edition, p. 51
- ↑ Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886-95. PMID 14985486.
- ↑ Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Begaud B. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 1996;335:609-16. PMID 8692238.
- ↑ Curnock AL, Dweik RA, Higgins BH, Saadi HF, Arroliga AC. High prevalence of hypothyroidism in patients with primary pulmonary hypertension. Am J Med Sci 1999;318:289-292. PMID 10555089.
- ↑ Cool CD, Rai PR, Yeager ME, Hernandez-Saavedra D, Serls AE, Bull TM, Geraci MW, Brown KK, Routes JM, Tuder RM, Voelkel NF. Expression of Human Herpesvirus 8 in Primary Pulmonary Hypertension.N Engl J Med 2003;349:1113-22. PMID 13679525.
- ↑ Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000;67:737-44. PMID 10903931.
- ↑ Blanpain C, Le Poul E, Parma J, Knoop C, Detheux M, Parmentier M, Vassart G, Abramowicz MJ. Serotonin 5-HT(2B) receptor loss of function mutation in a patient with fenfluramine-associated primary pulmonary hypertension. Cardiovasc Res 2003;60(3):518-28. PMID 14659797.
- ↑ Proceedings of the 3rd World Symposium on Pulmonary Arterial Hypertension. Venice, Italy, June 23-25, 2003. J Am Coll Cardiol 2004 Jun 16;43(12 Suppl S):1S-90S. PMID 15194171.
External links
- Goldminer: Pulmonary hypertension
- The Merck Manual of Diagnosis and Therapy: Pulmonary Hypertension
- The Pulmonary Hypertension Association
- PH Central - the internet resource for Pulmonary Arterial Hypertension
- Facts About Primary Pulmonary Hypertension from National Heart, Lung, and Blood Institute (NHLBI)
- Webcast: The Changing World of Pulmonary Arterial Hypertension Therapies - American College of CHEST Physicians
- pph at NIH/UW GeneTests - "BMPR2-Related Primary Pulmonary Hypertension"
de:Pulmonale Hypertonie it:Ipertensione arteriosa polmonare nl:Pulmonale hypertensie simple:Pulmonary hypertension uk:Легенева гіпертензія