Hypertrophic cardiomyopathy pathophysiology
|
Hypertrophic Cardiomyopathy Microchapters |
|
Differentiating Hypertrophic Cardiomyopathy from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Hypertrophic cardiomyopathy pathophysiology On the Web |
|
Directions to Hospitals Treating Hypertrophic cardiomyopathy |
|
Risk calculators and risk factors for Hypertrophic cardiomyopathy pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Soroush Seifirad, M.D.[2]
Overview
The progression to Hypertrophic cardiomyopathy usually involves the mutations in contractile sarcomeric proteins of myocardium, which describe the presence of left ventricular hypertrophy (LVH) in the absence of an increased external load (unexplained LVH). Additionally, HCM hypertrophy is generally asymmetric.
HCM is the most common genetically transmitted cardiovascular disease. Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins. Penetrance of HCM is incomplete, variable and time or age-related. The disease may be sporadic but affected family members are discovered in 13% of cases. More than 200 mutations involving at least 10 chromosomes encoding structural proteins of the myocyte have been discovered. These mutations have varying degrees of penetrance and even the same mutation may have variable expression, implying the superimposed effects of other genes or environmental influences. Children of a patient with HCM have a 50% chance of inheriting the trait.
Depending on the degree of obstruction of the outflow of blood from the left ventricle of the heart, HCM can be defined as obstructive or non-obstructive. About 25% of individuals with HCM demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals, obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction).
Although there may be structural or functional obstruction of the left ventricular outflow tract, symptoms may arise more often from diastolic dysfunction.There is extensive periarteriolar fibrosis that results in microvascular dysfunction and impairment in coronary flow reserve in patients with hypertrophic obstructive cardiomyopathy. Individuals with HCM have some degree of left ventricular hypertrophy. In approximately 2/3rds of cases this is asymmetric hypertrophy, involving the interventricular septum, and is known as asymmetric septal hypertrophy (ASH). This is in contrast to the symmetric and concentric hypertrophy seen in aortic stenosis or hypertension. On histopathologic examination, hypertrophic cardiomyopathy is characterized by both myocardial disarrays and by periarteriolar fibrosis. Myocardial disarray can be associated with aberrant impulse conduction and arrhythmias, and periarteriolar fibrosis can be associated with myocardial ischemia.
Pathophysiology
Physiology
The normal physiology of myocardium can be understood as follows:
- The myocardium is composed of specialized cardiac muscle cells with an ability not possessed by muscle tissue elsewhere in the body. Cardiac muscle, like other muscles, can contract, but it can also carry an action potential (i.e. conduct electricity), like the neurones that constitute nerves.
Pathogenesis
- The progression to Hypertrophic cardiomyopathy usually involves the mutations in contractile sarcomeric proteins of myocardium, which describe the presence of left ventricular hypertrophy (LVH) in the absence of an increased external load (unexplained LVH).
- Additionally, HCM hypertrophy is generally asymmetric.
Genetics
Hypertrophic cardiomyopathy is transmitted in an autosomal dominant pattern. Genes involved in the pathogenesis of Hypertrophic cardiomyopathy include:
- [Gene1]
- [Gene2]
- [Gene3]
The development of Hypertrophic cardiomyopathy is the result of multiple genetic mutations such as:
- [Mutation 1]
- [Mutation 2]
- [Mutation 3]
HCM is the most common genetically transmitted cardiovascular disease. Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15]. Penetrance of HCM is incomplete, variable and time or age-related. The disease may be sporadic but affected family members are discovered in 13% of cases. More than 200 mutations involving at least 10 chromosomes encoding structural proteins of the myocyte have been discovered. These mutations have varying degrees of penetrance and even the same mutation may have variable expression, implying superimposed effects of other genes or environmental influences. Children of a patient with HCM have a 50% chance of inheriting the trait.
Mutations
Common Mutations
Mutations in three regions affect more than half the patients with HCM:
- Beta-myosin heavy chain
- Myosin binding protein C
- Cardiac troponin T
Complete List of Mutations
Hypertrophic cardiomyopathy is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins including beta-cardiac myosin heavy chain (the first gene identified), cardiac actin, cardiac troponin T, alpha-tropomyosin, cardiac troponin I, cardiac myosin-binding protein C, and the myosin light chains. Specific gene mutations that have been identified include the following:
| Gene | Locus | Type |
|---|---|---|
| MYH7 | 14q12 | CMH1 |
| TNNT2 | 1q32 | CMH2 |
| TPM1 | 15q22.1 | CMH3 (115196) |
| MYBPC3 | 11p11.2 | CMH4 (115197) |
| ? | ? | CMH5 |
| PRKAG2 | 7q36 | CMH6 (600858) |
| TNNI3 | 19q13.4 | CMH7 |
| MYL3 | 3p | CMH8 (608751) |
| TTN | 2q24.3 | CMH9 |
| MYL2 | 12q23-q24 | CMH10 |
| ACTC1 | 15q14 | CMH11 (612098) |
| CSRP3 | 11p15.1 | CMH12 (612124) |
While the above table represents the most common genetic mutations, there are also about 200 intergenic (within a gene) mutations. These include missense and single amino acid residue substitutions. There are different genetic mutations in different families. The environment may also play a role because affected individuals in the same family may have a different phenotypic expression (i.e different degrees of left ventricular hypertrophy). The goal of modifier genes in regulating phenotypic expression is not clear.
While genes, gene modifiers, and environment may play a role in the phenotypic expression of left ventricular hypertrophy, genes may also play a role in the risk of arrhythmias. While most literature so far focuses on European, American, and Japanese populations, HCM appears in all racial groups. The incidence of HCM is about 0.2% to 0.5% of the general population.
Specific Chromosomal Abnormalities
β Myosin Heavy Chain-Chromosome 14 q11.2-3
In individuals without a family history of HCM, the most common cause of the disease is a de novo mutation of the gene that produces the β-myosin heavy chain. This chromosomal abnormality accounts for approximately 35%-45% of HCM cases. Significant LVH (left ventricular hypertrophy) is usually present. The Arg403Gln mutation is associated with an extremely poor prognosis with an average age of death at 33 years, while the Val606Met mutation is associated with a better prognosis.
Cardiac Troponin T-Chromosome 11
Accounts for approximately 15% of cases. Substantially less hypertrophy is noted but histology demonstrates the characteristic myocyte disarray of HCM. Most mutations of this gene are associated with markedly reduced survival.
Cardiac Myosin Binding Protein-C-Chromosome 11
This chromosomal abnormality accounts for 15% to 35% of patients, but given the reduced penetrance associated with this abnormality, the true incidence may actually be greater. Patients generally present later in life and in general, have a better prognosis than beta myosin heavy chain or cardiac troponin T mutations. Up to 60% of patients at age 50 years have no evidence of LVH. LVH may appear later in life in these patients. Because of this, a normal EKG and a normal ECHO at age 18 does not exclude the presence of HCM.
Arg663 His mutation
The beta-myosin heavy chain Arg663 His mutation is associated with a higher risk of atrial fibrillation [16].
PRKAG2 Mutation
There is no myocyte disarray, but the conduction block is present. This variant is more akin to a storage disease[17].
Mutations that Alter the Phenotypic Expression of the Disease
An insertion/deletion polymorphism in the gene encoding for angiotensin converting enzyme (ACE) alters the clinical phenotype of the disease. The D/D (deletion/deletion) genotype of ACE is associated with more marked hypertrophy of the left ventricle and may be associated with higher risk of adverse outcomes [18] [19].
Genetic Testing
Whenever a mutation is identified through genetic testing, family-specific genetic testing can be used to identify relatives at-risk for the disease (HCM Genetic Testing Overview). In individuals without a family history of HCM, the most common cause of the disease is a de novo mutation of the gene that produces the β-myosin heavy chain.
Outflow Obstruction
Depending on the degree of obstruction of the outflow of blood from the left ventricle of the heart, HCM can be defined as obstructive or non-obstructive. About 25% of individuals with HCM demonstrate an obstruction to the outflow of blood from the left ventricle during rest. In other individuals obstruction only occurs under certain conditions. This is known as dynamic outflow obstruction, because the degree of obstruction is variable and is dependent on the amount of blood in the ventricle immediately before ventricle systole (contraction).
Location Of The Left Ventricular Outflow Obstruction
The left ventricular obstruction can be either
- Mid-cavitary: the middle of the ventricle or
- Sub-aortic: just below the aortic valve
Classification of the Valve Gradient in Hypertrophic Cardiomyopathy
The valve gradient in HCM can be classified into three categories:
- A gradient greater than 30 mm Mercury under basal conditions
- A gradient that is greater than 30 mm Mercury with provocation
- A gradient that is less than 30 mm Mercury at rest and with provocation
Maneuvers that Increase the Outflow Gradient
- Amyl nitrite inhalation
- Valsalva maneuver
- Premature ventricular contractions (PVCs)
- Isoproterenol infusion
- Dobutamine infusion; but this is not recommended as a diagnostic tool[20][21]
- Treadmill or exercise stress testing
Causes of Left Ventricular Outflow Obstruction: Systolic Anterior Motion of the Mitral Valve (SAM)
If dynamic outflow obstruction is present in a patient with HCM, it is usually due to systolic anterior motion (SAM) of the anterior leaflet of the mitral valve. The systolic anterior motion of the mitral valve (SAM) may be due to a subaortic bulge of the septum along with narrowing the left ventricular outflow tract, which taken together cause high-velocity flow. This, in turn, is associated with the Venturi effect which is a local low-pressure zone in the left ventricular outflow tract. This low-pressure zone was thought to suck the mitral valve anteriorly into the septum. More recently, however, SAM onset has been observed to be instead a low-velocity phenomenon. [22] [23]. The role of Venturi forces in the left ventricular outflow tract may be less important than previously thought. While the Venturi effect was thought to cause the abnormality in prior studies, more recent echocardiographic studies indicates that drag, which is more of a pushing force rather than a sucking force like the Venturi effect, maybe the dominant hydrodynamic force acting on the mitral leaflets [22][23][24][25][26] [27].
The videos below show examples of systolic anterior motion of the mitral valve:
{{#ev:youtube|Y7JUVTXHBs0}}
{{#ev:youtube|6TWb-wIL0H0}}
Impact of Systolic Anterior Motion of the Mitral Valve: The Spike and Dome Pattern to the Carotid Pulse
Because the mitral valve leaflet doesn't get pulled into the left ventricular outflow tract (LVOT) until after the aortic valve opens, the initial upstroke of the arterial pulse pressure will be normal. When the mitral valve leaflet gets pushed into the LVOT, the arterial pulse will momentarily collapse and will later be followed by a second rise in the pulse pressure, as the left ventricular pressure overcomes the increased obstruction caused by the SAM of the mitral valve. This can be seen on the physical examination as a double-tap upon palpation of the apical impulse and as a double pulsation upon palpation of the carotid pulse, known as pulsus bisferiens or a "spike and dome pattern" to the carotid pulse.
Accompanying Mitral Regurgitation
As a result of the drag effect or the Venturi effect, there may be mild to moderate mitral regurgitation in association with hypertrophic cardiomyopathy. Most often the mitral regurgitation jet is directed posteriorly. If the jet is not directed posteriorly then other diagnoses should be considered which include myxomatous degeneration or other anomalies of the mitral valve.
Pathophysiologic Consequences of Outflow Obstruction
Chronic outflow obstruction and result in the following abnormalities[28][29]:
- Increased left ventricular wall stress
- Myocardial ischemia
- Myocardial necrosis
- Replacement fibrosis
Prognostic Significance of Outflow Obstruction
The presence of outflow obstruction is associated with a twofold increased risk of death and a 4.4 fold increase in the risk of progression to New York Heart Association class III or IV heart failure [30][31]. Above a gradient of 30 mm Hg, there was no further increase in the risk of sudden cardiac death or progression of congestive heart failure symptoms[32].
Ischemia
There is extensive periarteriolar fibrosis that results in microvascular dysfunction and impairment in coronary flow reserve in patients with hypertrophic obstructive cardiomyopathy.
Histopathology
Compared to normal arterioles on the left, the arterioles from a patient with hyertension (middle) show moderate periarteriolar thickening and fibrosis. Shown on the right is a patient with HCM in which there is even more signficant periarteriolar thickening and fibrosis. This thickening of the wall of the intramyocardial arterioles leads to an increased wall/lumen ratio, subendocardial ischemia and impaired coronary flow reserve[33][34]. Patients who subsequently died in one series had abnormal coronary flow reserve on PET scanning at baseline indicating that ischemia may play a role, at least in part, in subsequent mortality.
-
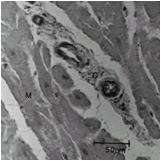
Normal arteriole
-
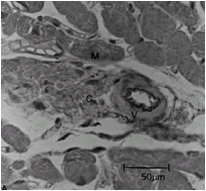
Hypertensive arteriole with wall thickening and myocyte hypertrophy
-
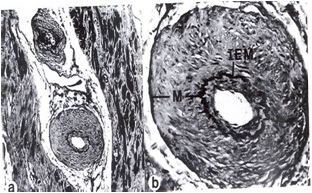
Arteriole in HCM patient with periarteriole fibrosis and thicknening



Arrhythmogenesis
Patients with hypertrophic cardiomyopathy are at risk of arrhythmias and sudden death. Abnormal filling of the left atrium may result in the left atrial dilation which may predispose the patient to atrial fibrillation. The presence of myocardial disarray and myocardial ischemia (due to microvascular dysfunction and episodes of reduced cardiac output) may predispose the patient to ventricular tachycardia, ventricular fibrillation, and sudden cardiac death.
Atrial Arrhythmias
Impaired filling of the left ventricle can lead to left atrial stretch and left atrial dilation. This, in turn, can predispose the patient to the development of atrial fibrillation. The onset of atrial fibrillation can be quite dangerous in these patients as the loss of left atrial kick and the more rapid heart rate can both diminish left ventricular filling which can lead to severe hemodynamic compromise. This hemodynamic compromise can, in turn, be associated with sudden cardiac death.
Ventricular Arrhythmias
Ventricular arrhythmias and degeneration into sudden cardiac death may be due to the following:
- Primary arrhythmias
- The presence of myocardial disarray
- The presence of scar or fibrosis
- Hemodynamic instability with diminished stroke volume
- The presence of ischemia
It must be emphasized that atrial arrhythmias (which are commonly detected on ambulatory monitoring) can lead to ischemia and hemodynamic compromise which may, in turn, lead to sudden cardiac death in these patients as well.
Autonomic Imbalance
Assessment of autonomic function in patients with HCM often reveals abnormal responses of heart rate and blood pressure to exercise in two-thirds, which was associated with a more malignant clinical course, suggesting that autonomic imbalance may also be important in the genesis of sudden cardiac death in these patients.
Anatomic abnormalities
Individuals with HCM have some degree of left ventricular hypertrophy. In approximately 2/3rds of cases this is asymmetric hypertrophy, involving the interventricular septum, and is known as asymmetric septal hypertrophy (ASH). This is in contrast to the symmetric and concentric hypertrophy seen in aortic stenosis or hypertension.
Left Ventricle
The degree of ventricular hypertrophy is variable ranging from diffuse involvement of both ventricles to isolated involvement of a portion of one segment of the LV.
Data from two large registries indicate that;
- 55% of cases involve the septum and anterolateral free wall,
- 20% involve the entire septum alone,
- 10% is limited to the nasal septum and 15% are limited to the apical or distal LV (Yamaguchi variant).
Some genetic variants may manifest very little overt LVH but are still associated with an increased risk of sudden cardiac death (SCD).
Outflow Tract
The left ventricular outflow tract is often small.
Mitral Valve
The mitral valve maybe elongated and enlarged.
Associated Conditions
Conditions associated with Hypertrophic cardiomyopathy include:
- [Condition 1]
- [Condition 2]
- [Condition 3]
Histopathologic Abnormalities
On histopathologic examination, hypertrophic cardiomyopathy is characterized by both myocardial disarrays and by periarteriolar fibrosis. Myocardial disarray can be associated with aberrant impulse conduction and arrhythmias, and periarteriolar fibrosis can be associated with myocardial ischemia.
Myocardial Disarray
In HCM, the normal alignment of muscle cells is disrupted (there is a swirling pattern to the arrangement of the muscle cells), a phenomenon known as myocardial disarray. HCM is believed to be due to a mutation in one of many genes that results in a mutated myosin heavy chain, one of the components of the myocyte (the muscle cell of the heart). Histopathologically, the cardiac sarcomere is abnormal resulting in hypertrophy of the left ventricle in the absence of other disorders that could produce the condition such as hypertension, amyloid or aortic stenosis. The presence of myocardial disarray may be associated with abnormalities of electrical conduction in the heart (including electrical reentry loops) which thereby contributes to an increased risk of sudden cardiac death.
-
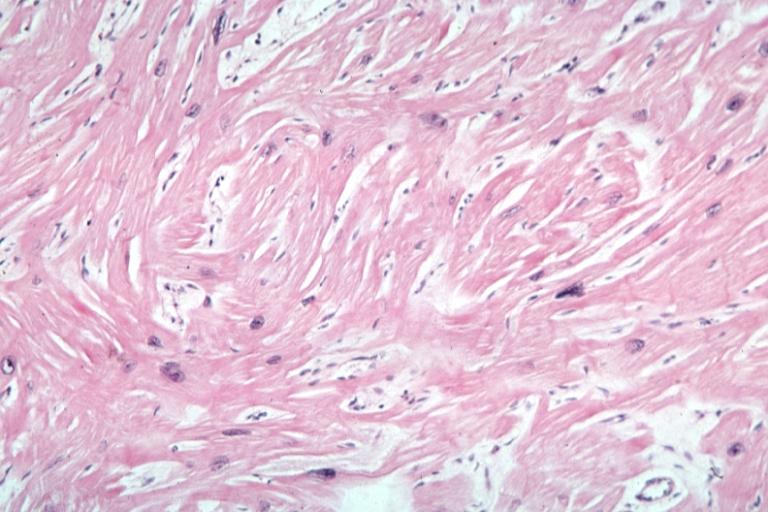
Myocardial disarray with swirling pattern of myocytes
-
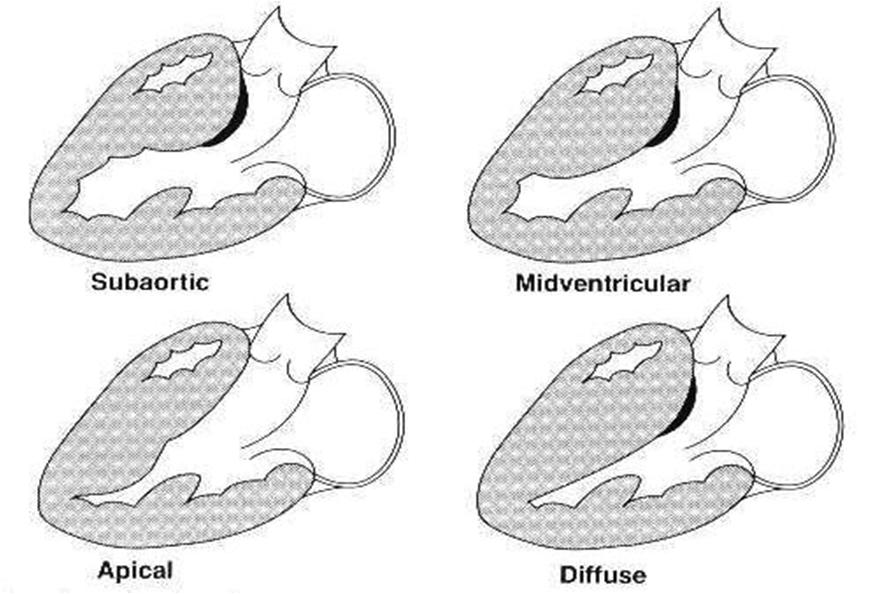
Variants of hypertrophic cardiomyopathy
-
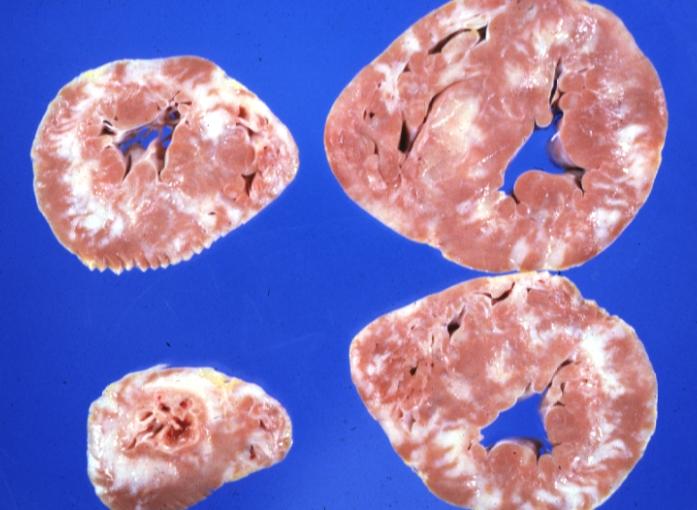
White areas of fibrosis or scar in a patient with HCM which may contribute in part to arrhythmias



Periarteriolar Fibrosis
Compared to normal arterioles on the left, the arterioles from a patient with hyertension (middle) show moderate periarteriolar thickening and fibrosis. Shown on the right is a patient with HCM in which there is even more signficant periarteriolar thickening and fibrosis. This thickening of the wall of the intramyocardial arterioles leads to an increased wall/lumen ratio, subendocardial ischemia and impaired coronary flow reserve[33][34]. Patients who subsequently died in one series had abnormal coronary flow reserve on PET scanning at baseline indicating that ischemia may play a role, at least in part, in subsequent mortality.
-
Normal arteriole
-
Hypertensive arteriole with wall thickening and myocyte hypertrophy
-
Arteriole in HCM patient with periarteriole fibrosis and thicknening
Gross Pathology
On gross pathology, asymmetric interventricular wall thickening is characteristic findings of hypertrophic cardiomyopathy. Subaortic stenosis could be evident in many cases. In the Yamaguchi subtype, there is apical hypertrophy.
Microscopic Pathology
On microscopic histopathological analysis, myocardial disarray, periarteriolar fibrosis, and hypertrophy are characteristic findings of hypertrophic cardiomyopathy.
Histopathologically, small vessels have hypertrophy of the tunica media. Combined with increased wall tension, decreased vasodilator reserve, and inadequate capillary density, there is a mismatch between blood supply and demand. Over time, it is thought that there is repeated ischemia followed by fibrosis and eventually, dilation and systolic dysfunction (“burned out hypertrophy”).
-
Micro med mag H&E mid-mural myocardium with hypertrophy and interstitial fibrosis atrophy is present marked increase in interstitial fibroblastic cells
-
Micro high mag H&E myofiber hypertrophy and interstitial fibrosis with marked increase in interstitial fibroblastic cells


-
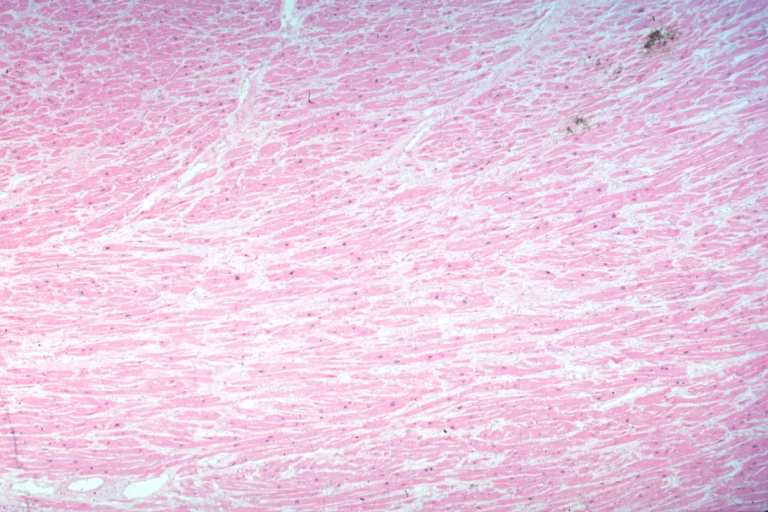
Micro med mag H&E myofiber hypertrophy some atrophy interstitial fibrosis with many fibroblastic cells
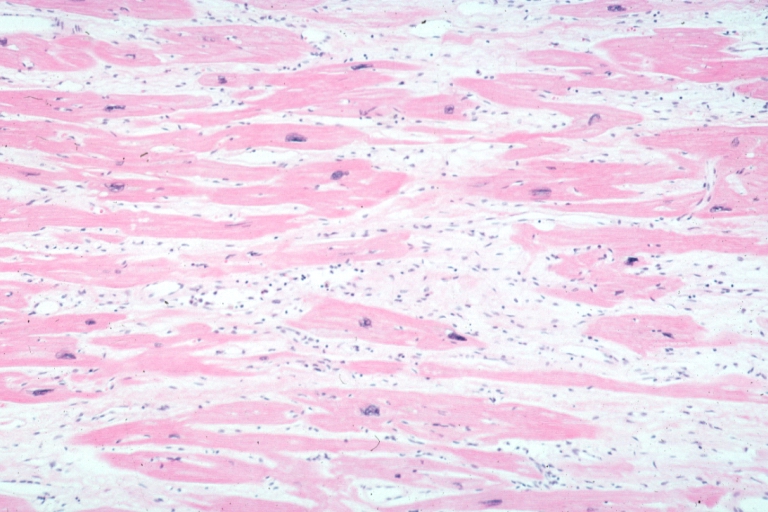
-
Micro high mag H&E hypertrophied fibers with some evidence of atrophy and marked interstitial fibrosis with many fibroblastic type cells


-
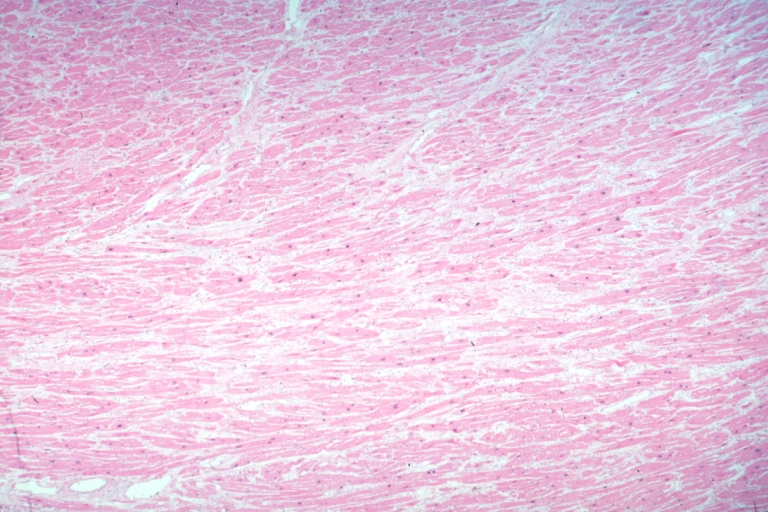
Micro low mag H&E shows myofiber hypertrophy and interstitial fibrosis
-
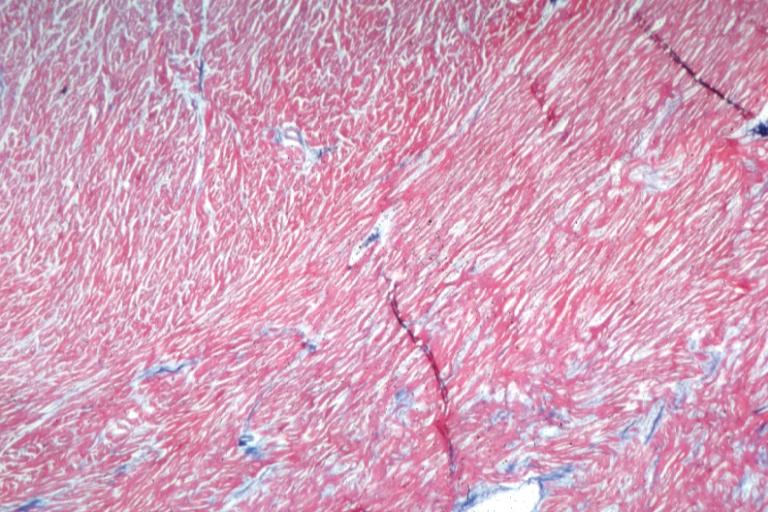
Cardiomyopathy: Micro H&E low mag interventricular septum at junction of normal myofiber orientation with asymmetrical hypertrophy (an excellent example)


-
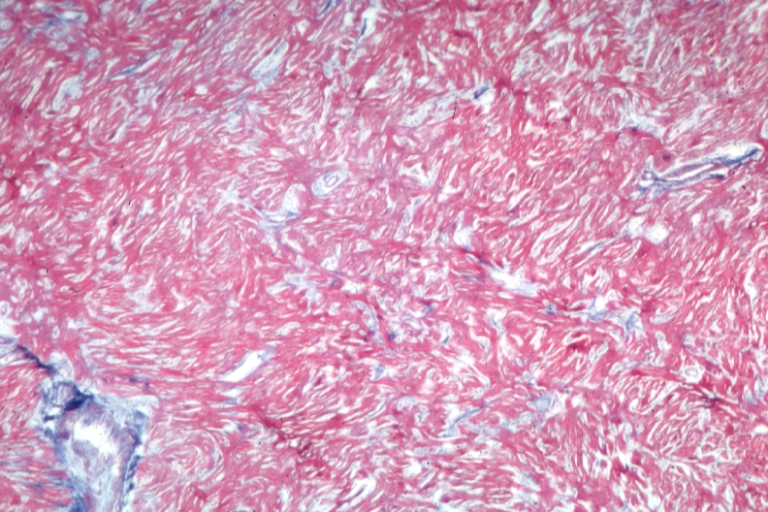
Cardiomyopathy: Micro H&E low mag marked myofiber disarray asymmetrical hypertrophy
-
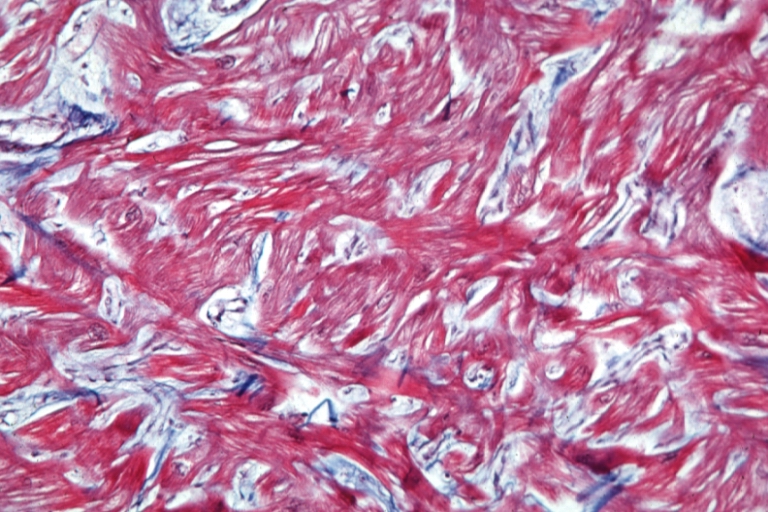
Cardiomyopathy: Micro trichrome high mag marked myofiber disarray


-
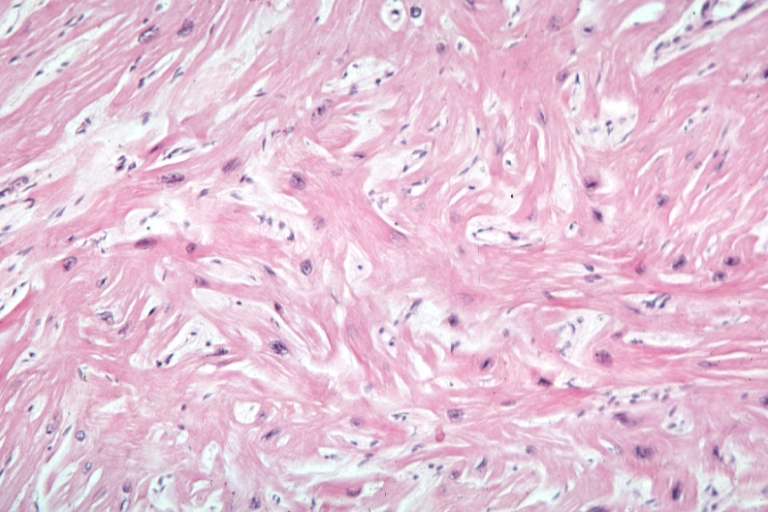
Cardiomyopathy: Micro H&E med mag excellent example myofiber disarray
-
Cardiomyopathy: Micro H&E high mag excellent example myofiber disarray

References
- ↑ Maron BJ, Moller JH, Seidman CE et al. Impact of laboratory molecular diagnosis on contemporary diagnostic criteria for genetically transmitted cardiovascular diseases. Hypertrophic cardiomyopathy, long-QT syndrome, and Marfan syndrome. [A statement for healthcare professionals from the Councils on Clinical Cardiology, Cardiovascular Disease in the Young, and Basic Science, American Heart Association]. Circulation 1998;98:1460–71.
- ↑ Schwartz K, Carrier L, Guicheney P, Komajda M. Molecular basis of familial cardiomyopathies. Circulation 1995;91:532–40.
- ↑ Niimura H, Bachinski LL, Sangwatanaroj S et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998;338:1248–57.
- ↑ Thierfelder L, Watkins H, MacRae C et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy. A disease of the sarcomere. Cell 1994;77:701–12.
- ↑ Watkins H, McKenna WJ, Thierfelder L et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 1995;332:1058–64.
- ↑ Charron P, Dubourg O, Desnos M et al. Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation 1998;97: 2230–6.
- ↑ Maron BJ, Niimura H, Casey SA et al. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J Am Coll Cardiol 2001;38:315–21.
- ↑ Anan R, Greve G, Thierfelder L et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest 1994;93:280–5.
- ↑ Coviello DA, Maron BJ, Spirito P et al. Clinical features of hypertrophic cardiomyopathy caused by mutation of a “hot spot” in the alpha-tropomyosin gene. J Am Coll Cardiol 1997;29:635–40.
- ↑ Blair E, Redwood C, Ashrafian H et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy. Evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001;10:1215–20.
- ↑ Erdmann J, Raible J, Maki-Abadi J et al. Spectrum of clinical phenotypes and gene variants in cardiac myosin-binding protein C mutation carriers with hypertrophic cardiomyopathy. J Am Coll Cardiol 2001;38:322–30.
- ↑ Gruver EJ, Fatkin D, Dodds GA et al. Familial hypertrophic cardiomyopathy and atrial fibrillation caused by Arg663His beta-cardiac myosin heavy chain mutation. Am J Cardiol 1999;83:13H–8H.
- ↑ Kimura A, Harada H, Park JE et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997;16:379–82.
- ↑ Marian AJ, Roberts R. Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 1995;92:1336–47.
- ↑ Niimura H, Patton KK, McKenna WJ et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002;105:446–51.
- ↑ Seidman JG, Seidman CE. The genetic basis for cardiomyopathy. From mutation identification to mechanistic paradigms. Cell 2001; 104:557–67.
- ↑ Arad M, Benson DW, Perez-Atayde AR et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 2002;109:357–62.
- ↑ Doolan G, Nguyen L, Chung J, Ingles J, Semsarian C. Progression of left ventricular hypertrophy and the angiotensin-converting enzyme gene polymorphism in hypertrophic cardiomyopathy. Int J Cardiol. 2004 Aug; 96(2):157–63. (Medline abstract)
- ↑ Marian AJ, Yu QT, Workman R, Greve G, Roberts R. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet. 1993 Oct 30; 342(8879):1085–6. (Medline abstract)
- ↑ Pellikka PA, Oh JK, Bailey KR, Nichols BA, Monahan KH, Tajik AJ. Dynamic intraventricular obstruction during dobutamine stress echocardiography. A new observation. Circulation 1992;86:1429–32.
- ↑ Okeie K, Shimizu M, Yoshio H et al. Left ventricular systolic dysfunction during exercise and dobutamine stress in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2000;36:856–63.
- ↑ 22.0 22.1 Jiang L, Levine RA, King ME, Weyman AE. An integrated mechanism for the systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy based on echocardiographic observations. Am Heart J 1987; 113:633–44
- ↑ 23.0 23.1 Sherrid MV, Gunsburg DZ, Moldenhauer S, Pearle G. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2000; 36:1344–54
- ↑ Sherrid MV, Chu Ck, DeLia E, Mogtader A, Dwyer Jr. EM, An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22:816–25
- ↑ Levine RA, Vlahakes GJ, Lefebvre X, et al. Papillary muscle displacement causes systolic anterior motion of the mitral valve. Circulation 1995; 91:1189–95
- ↑ Messmer BJ. Extended myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 1994; 58:575–7
- ↑ Schoendube FA, Klues HG, Reith S, Flachskampf FA, Hanrath P, Messmer BJ. Long-term clinical and echocardiographic follow-up after surgical correction of hypertrophic obstructive cardiomyopathy with extended myectomy and reconstruction of the subvalvular mitral apparatus. Circulation 1995; 92:II-122–7
- ↑ Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295–303.
- ↑ Choudhury L, Mahrholdt H, Wagner A et al. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002;40:2156–64.
- ↑ Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295–303.
- ↑ Kofflard MJ, Ten Cate FJ, van der Lee C, van Domburg RT. Hypertrophic cardiomyopathy in a large community-based population. Clinical outcome and identification of risk factors for sudden cardiac death and clinical deterioration. J Am Coll Cardiol 2003;41:987–93.
- ↑ Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295–303.
- ↑ 33.0 33.1 Lorenzoni R, Gistri R, Cecchi F, Olivotto I, Chiriatti G, Elliott P; et al. (1998). "Coronary vasodilator reserve is impaired in patients with hypertrophic cardiomyopathy and left ventricular dysfunction". Am Heart J. 136 (6): 972–81. PMID 9842009.
- ↑ 34.0 34.1 Choudhury L, Elliott P, Rimoldi O, Ryan M, Lammertsma AA, Boyd H; et al. (1999). "Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment". Basic Res Cardiol. 94 (1): 49–59. PMID 10097830.