Daratumumab
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Allison Tu [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Daratumumab is an antineoplastic agent that is FDA approved for the treatment of multiple myeloma. Common adverse reactions include fatigue, headache, nausea, diarrhea, constipation, decreased appetite, vomiting, lymphocytopenia, neutropenia, thrombocytopenia, anemia, back pain, arthralgia, leg pain, musculoskeletal chest pain, cough, nasal congestion, dyspnea, nasopharyngitis, pneumonia, and infusion-related reaction.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Daratumumab is indicated for, in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone, treatment of patients with multiple myeloma who have received at least one prior therapy; for, in combination with pomalidomide and dexamethasone, treatment of patients with multiple myeloma who have received at least two prior therapies including lenalidomide and a proteasome inhibitor; and as monotherapy, for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory agent or who are double refractory to a PI and an immunomodulatory agent.
Dosing Information
- The recommended dose of daratumumab for monotherapy and combination therapy with lenalidomide or pomalidomide and low-dose dexamethasone (4-week cycle regimens) is 16 mg/kg actual body weight administered as an intravenous infusion according to the following dosing schedule:
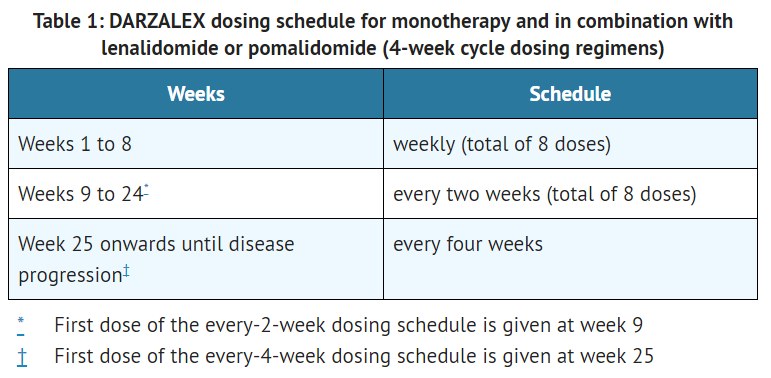
This image is provided by the National Library of Medicine. - The recommended dose of daratumumab for combination therapy with bortezomib and dexamethasone (3-week cycle regimen) is 16 mg/kg actual body weight administered as an intravenous infusion according to the following dosing schedule:
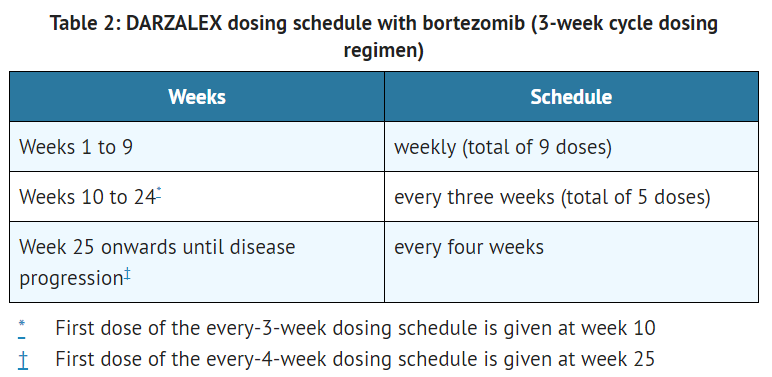
This image is provided by the National Library of Medicine.


Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Daratumumab in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Daratumumab in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding indications and dosing of daratumumab in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Daratumumab in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Daratumumab in pediatric patients.
Contraindications
There is limited information regarding contraindications of daratumumab.
Warnings
- Infusion Reactions
- Daratumumab can cause severe infusion reactions. Approximately half of all patients experienced a reaction, most during the first infusion. Infusion reactions can also occur with subsequent infusions. Nearly all reactions occurred during infusion or within 4 hours of completing daratumumab. Prior to the introduction of post-infusion medication in clinical trials, infusion reactions occurred up to 48 hours after infusion.
- Severe reactions have occurred, including bronchospasm, hypoxia, dyspnea, hypertension, laryngeal edema and pulmonary edema. Signs and symptoms may include respiratory symptoms, such as nasal congestion, cough, throat irritation, as well as chills, vomiting and nausea. Less common symptoms were wheezing, allergic rhinitis, pyrexia, chest discomfort, pruritus, and hypotension.
- Pre-medicate patients with antihistamines, antipyretics and corticosteroids. Frequently monitor patients during the entire infusion. Interrupt daratumumab infusion for reactions of any severity and institute medical management as needed. Permanently discontinue daratumumab therapy for life-threatening (Grade 4) reactions. For patients with Grade 1, 2, or 3 reactions, reduce the infusion rate when re-starting the infusion.
- To reduce the risk of delayed infusion reactions, administer oral corticosteroids to all patients following daratumumab infusions. Patients with a history of chronic obstructive pulmonary disease may require additional post-infusion medications to manage respiratory complications. Consider prescribing short- and long-acting bronchodilators and inhaled corticosteroids for patients with chronic obstructive pulmonary disease.
- Interference with serological testing
- Daratumumab binds to CD38 on red blood cells (RBCs) and results in a positive Indirect Antiglobulin Test (Indirect Coombs test). Daratumumab-mediated positive indirect antiglobulin test may persist for up to 6 months after the last daratumumab infusion. Daratumumab bound to RBCs masks detection of antibodies to minor antigens in the patient's serum. The determination of a patient's ABO and Rh blood type are not impacted.
- Notify blood transfusion centers of this interference with serological testing and inform blood banks that a patient has received daratumumab. Type and screen patients prior to starting daratumumab.
- Neutropenia
- Daratumumab may increase neutropenia induced by background therapy. Monitor complete blood cell counts periodically during treatment according to manufacturer's prescribing information for background therapies. Monitor patients with neutropenia for signs of infection. Daratumumab dose delay may be required to allow recovery of neutrophils. No dose reduction of daratumumab is recommended. Consider supportive care with growth factors.
- Thrombocytopenia
- Daratumumab may increase thrombocytopenia induced by background therapy. Monitor complete blood cell counts periodically during treatment according to manufacturer's prescribing information for background therapies. Daratumumab dose delay may be required to allow recovery of platelets. No dose reduction of daratumumab is recommended. Consider supportive care with transfusions.
- Interference with determination of complete response
- Daratumumab is a human IgG kappa monoclonal antibody that can be detected on both the serum protein electrophoresis (SPE) and immunofixation (IFE) assays used for the clinical monitoring of endogenous M-protein. This interference can impact the determination of complete response of disease progression in some patients with IgG kappa myeloma protein.
Adverse Reactions
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The safety data described below reflects exposure to daratumumab (16 mg/kg) in 820 patients with multiple myeloma including 526 patients from two Phase 3 active-controlled trials who received daratumumab in combination with either lenalidomide (DRd, n=283; Study 3) or bortezomib (DVd, n=243; Study 4) and five open-label, clinical trials in which patients received daratumumab either in combination with pomalidomide (DPd, n=103; Study 5), in combination with lenalidomide (n=35), or as monotherapy (n=156).
Combination treatment with lenalidomide: Adverse reactions described in Table 4 reflect exposure to daratumumab (DRd arm) for a median treatment duration of 13.1 months (range: 0 to 20.7 months) and median treatment duration of 12.3 months (range: 0.2 to 20.1 months) for the lenalidomide group (Rd) in Study 3. The most frequent adverse reactions (≥20%) were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasms, cough and dyspnea. The overall incidence of serious adverse reactions was 49% for the DRd group compared with 42% for the Rd group. Serious adverse reactions with at least a 2% greater incidence in the DRd arm compared to the Rd arm were pneumonia (12% vs Rd 10%), upper respiratory tract infection (7% vs Rd 4%), influenza and pyrexia (DRd 3% vs Rd 1% for each). Adverse reactions resulted in discontinuations for 7% (n=19) of patients in the DRd arm versus 8% (n=22) in the Rd arm.

Laboratory abnormalities worsening during treatment from baseline listed in Table 5.

Combination treatment with Bortezomib: Adverse reactions described in Table 6 reflect exposure to daratumumab (DVd arm) for a median treatment duration of 6.5 months (range: 0 to 14.8 months) and median treatment duration of 5.2 months (range: 0.2 to 8.0 months) for the bortezomib group (Vd) in Study 4. The most frequent adverse reactions (>20%) were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough and dyspnea. The overall incidence of serious adverse reactions was 42% for the DVd group compared with 34% for the Vd group. Serious adverse reactions with at least a 2% greater incidence in the DVd arm compared to the Vd arm were upper respiratory tract infection (DVd 5% vs Vd 2%), diarrhea and atrial fibrillation (DVd 2% vs Vd 0% for each). Adverse reactions resulted in discontinuations for 7% (n=18) of patients in the DVd arm versus 9% (n=22) in the Vd arm.

Laboratory abnormalities worsening during treatment are listed in Table 7.

Combination treatment with Pomalidomide: Adverse reactions described in Table 8 reflect exposure to daratumumab, pomalidomide and dexamethasone (DPd) for a median treatment duration of 6 months (range: 0.03 to 16.9 months) in Study 5. The most frequent adverse reactions (>20%) were infusion reactions, diarrhea, constipation, nausea, vomiting, fatigue, pyrexia, upper respiratory tract infection, muscle spasms, back pain, arthralgia, dizziness, insomnia, cough and dyspnea. The overall incidence of serious adverse reactions was 49%. Serious adverse reactions reported in ≥5% patients included pneumonia (7%). Adverse reactions resulted in discontinuations for 13% of patients.

Laboratory abnormalities worsening during treatment are listed in Table 9.

Monotherapy: The safety data reflect exposure to daratumumab in 156 adult patients with relapsed and refractory multiple myeloma treated with daratumumab at 16 mg/kg in three open-label, clinical trials. The median duration of exposure was 3.3 months (range: 0.03 to 20.04 months). Serious adverse reactions were reported in 51 (33%) patients. The most frequent serious adverse reactions were pneumonia (6%), general physical health deterioration (3%), and pyrexia (3%). Adverse reactions resulted in treatment delay for 24 (15%) patients, most frequently for infections. Adverse reactions resulted in discontinuations for 6 (4%) patients. Adverse reactions occurring in at least 10% of patients are presented in Table 10. Table 11 describes Grade 3–4 laboratory abnormalities reported at a rate of ≥10%.


Infusion Reactions: In clinical trials (monotherapy and combination treatment; N=820) the incidence of any grade infusion reactions was 46% with the first infusion of daratumumab, 2% with the second infusion, and 3% with subsequent infusions. Less than 1% of patients had a Grade 3 infusion reaction with second or subsequent infusions. The median time to onset of a reaction was 1.4 hours (range: 0.02 to 72.8 hours). The incidence of infusion modification due to reactions was 42%. Median durations of infusion for the 1st, 2nd and subsequent infusions were 7.0, 4.3, and 3.5 hours respectively. Severe (Grade 3) infusion reactions included bronchospasm, dyspnea, laryngeal edema, pulmonary edema, hypoxia, and hypertension. Other adverse infusion reactions (any Grade, ≥5%) were nasal congestion, cough, chills, throat irritation, vomiting and nausea.
Herpes Zoster Virus Reactivation: Prophylaxis for Herpes Zoster Virus reactivation was recommended for patients in some clinical trials of daratumumab. In monotherapy studies, herpes zoster was reported in 3% of patients. In the randomized controlled combination therapy studies, herpes zoster was reported in 2% each in the DRd and Rd groups respectively (Study 3), in 5% versus 3% in the DVd and Vd groups respectively (Study 4) and in 2% of patients receiving DPd (Study 5).
Infections: In patients receiving daratumumab combination therapy, Grade 3 or 4 infections were reported with daratumumab combinations and background therapies (DVd: 21%, Vd: 19%; DRd: 28%, Rd: 23%; DPd: 28%). Pneumonia was the most commonly reported severe (Grade 3 or 4) infection across studies. Discontinuations from treatment were reported in 3% versus 2% of patients in the DRd and Rd groups respectively, 4% versus 3% of patients in the DVd and Vd groups respectively and in 5% of patients receiving DPd. Fatal infections were reported in 0.8% to 2% of patients across studies, primarily due to pneumonia and sepsis.
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. In clinical trials of patients with multiple myeloma treated with daratumumab as monotherapy or as combination therapies, none of the 111 evaluable monotherapy patients, and 2 (0.7%) of the 298 combination therapy patients, tested positive for anti-daratumumab antibodies. One patient administered daratumumab as combination therapy, developed transient neutralizing antibodies against daratumumab. However, this assay has limitations in detecting anti-daratumumab antibodies in the presence of high concentrations of daratumumab; therefore, the incidence of antibody development might not have been reliably determined. Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication and the underlying disease. Therefore, comparison of the incidence of antibodies to daratumumab with the incidence of antibodies to other products may be misleading.
Adverse reactions by organ system:
- Central Nervous System: Fatigue, headache, chills
- Cardiovascular: Hypertension
- Respiratory: Cough, nasal congestion, dyspnea, nasopharyngitis, pneumonia
- Gastrointestinal: Nausea, diarrhea, constipation, decreased appetite, vomiting
- Hematologic & oncologic: Lymphocytopenia, neutropenia, thrombocytopenia, anemia
- Infection: Herpes zoster
- Neuromuscular & skeletal: Back pain, arthralgia, leg pain, musculoskeletal chest pain
- Miscellaneous: Infusion-related reaction, fever, physical health deterioration
Postmarketing Experience
There is limited information regarding Daratumumab Postmarketing Experience in the drug label.
Drug Interactions
Indirect Antiglobulin Tests
Daratumumab binds to CD38 on RBCs and interferes with compatibility testing, including antibody screening and cross matching. Daratumumab interference mitigation methods include treating reagent RBCs with dithiothreitol (DTT) to disrupt daratumumab binding or genotyping. Since the Kell blood group system is also sensitive to DTT treatment, K-negative units should be supplied after ruling out or identifying alloantibodies using DTT-treated RBCs.If an emergency transfusion is required, non-cross-matched ABO/RhD-compatible RBCs can be given per local blood bank practices.
Serum Protein Electrophoresis and Immunofixation Tests
Daratumumab may be detected on serum protein electrophoresis (SPE) and immunofixation (IFE) assays used for monitoring disease monoclonal immunoglobulins (M protein). This can lead to false positive SPE and IFE assay results for patients with IgG kappa myeloma protein impacting initial assessment of complete responses by International Myeloma Working Group (IMWG) criteria. In patients with persistent very good partial response, consider other methods to evaluate the depth of response.
Use in Specific Populations
Pregnancy
Pregnancy Category (FDA):
There are no human data to inform a risk with use of daratumumab during pregnancy. Animal studies have not been conducted. However, there are clinical considerations. Immunoglobulin G1 (IgG1) monoclonal antibodies are transferred across the placenta. Based on its mechanism of action, daratumumab may cause fetal myeloid or lymphoid-cell depletion and decreased bone density. Defer administering live vaccines to neonates and infants exposed to daratumumab in utero until a hematology evaluation is completed. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively. Mice that were genetically modified to eliminate all CD38 expression (CD38 knockout mice) had reduced bone density at birth that recovered by 5 months of age. In cynomolgus monkeys exposed during pregnancy to other monoclonal antibodies that affect leukocyte populations, infant monkeys had a reversible reduction in leukocytes.
Pregnancy Category (AUS):
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Daratumumab in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Daratumumab during labor and delivery.
Nursing Mothers
There is no information regarding the presence of daratumumab in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG is known to be present in human milk. Published data suggest that antibodies in breast milk do not enter the neonatal and infant circulations in substantial amounts. The developmental and health benefits of breast-feeding should be considered along with the mother's clinical need for daratumumab and any potential adverse effects on the breast-fed child from daratumumab or from the underlying maternal condition.
Pediatric Use
There is no FDA guidance on the use of Daratumumab in pediatric settings.
Geriatic Use
Of the 156 patients that received daratumumab monotherapy at the recommended dose, 45% were 65 years of age or older, and 10% were 75 years of age or older. Of 664 patients that received daratumumab with various combination therapies, 41% were 65 to 75 years of age, and 9% were 75 years of age or older. No overall differences in safety or effectiveness were observed between these patients and younger patients.
Gender
There is no FDA guidance on the use of Daratumumab with respect to specific gender populations.
Race
There is no FDA guidance on the use of Daratumumab with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Daratumumab in patients with renal impairment.
Hepatic Impairment
There is no FDA guidance on the use of Daratumumab in patients with hepatic impairment.
Females of Reproductive Potential and Males
To avoid exposure to the fetus, women of reproductive potential should use effective contraception during treatment and for 3 months after cessation of daratumumab treatment.
Immunocompromised Patients
There is no FDA guidance one the use of Daratumumab in patients who are immunocompromised.
Administration and Monitoring
Administration
- Administer pre-infusion medication to reduce the risk of delayed infusion reactions to all patients 1-3 hours prior to every infusion of daratumumab as follows:
- Administer corticosteroids (monotherapy: methylprednisolone 100 mg, or equivalent), administered intravenously. Following the second infusion, the dose of corticosteroid may be reduced (oral or intravenous methylprednisolone 60 mg) or combination therapy: administer 20 mg dexamethasone prior to every daratumumab infusion. Dexamethasone is given intravenously prior to the first daratumumab infusion and oral administration may be considered prior to subsequent infusions).
- Antipyretics (oral acetaminophen 650 to 1000 mg).
- Antihistamine (oral or intravenous diphenhydramine 25 to 50 mg or equivalent) to reduce the risk of infusion reactions to all patients 1–3 hours prior to every infusion of daratumumab.
- Administer post-infusion medication to reduce the risk of delayed infusion reactions to all patients as follows:
- Monotherapy: Administer oral corticosteroid (20 mg methylprednisolone or equivalent dose of an intermediate-acting or long-acting corticosteroid in accordance with local standards) on each of the 2 days following all daratumumab infusions (beginning the day after the infusion).
- Combination therapy: Consider administering low-dose oral methylprednisolone (≤ 20 mg) or equivalent, the day after the daratumumab infusion. However, if a background regimen-specific corticosteroid (e.g. dexamethasone) is administered the day after the daratumumab infusion, additional post-infusion medications may not be needed.
- In addition, for any patients with a history of chronic obstructive pulmonary disease, consider prescribing post-infusion medications such as short and long-acting bronchodilators, and inhaled corticosteroids. Following the first four infusions, if the patient experiences no major infusion reactions, these additional inhaled post-infusion medications may be discontinued.
- Initiate antiviral prophylaxis to prevent herpes zoster reactivation within 1 week after starting daratumumab and continue for 3 months following treatment.
- Administer only as an intravenous infusion after dilution in 0.9% Sodium Chloride Injection, USP. Daratumumab should be administered by a healthcare professional, with immediate access to emergency equipment and appropriate medical support to manage infusion reactions if they occur.
- If a planned dose of daratumumab is missed, administer the dose as soon as possible and adjust the dosing schedule accordingly, maintaining the treatment interval.
- Administer daratumumab infusion intravenously at the infusion rate described below. Consider incremental escalation of the infusion rate only in the absence of infusion reactions.

This image is provided by the National Library of Medicine.
- For infusion reactions of any grade/severity, immediately interrupt the daratumumab infusion and manage symptoms. Management of infusion reactions may further require reduction in the rate of infusion, or treatment discontinuation of daratumumab as outlined below:
- Grade 1–2 (mild to moderate): Once reaction symptoms resolve, resume the infusion at no more than half the rate at which the reaction occurred. If the patient does not experience any further reaction symptoms, infusion rate escalation may resume at increments and intervals as clinically appropriate up to the maximum rate of 200 mL/hour (Table 3).
- Grade 3 (severe): Once reaction symptoms resolve, consider restarting the infusion at no more than half the rate at which the reaction occurred. If the patient does not experience additional symptoms, resume infusion rate escalation at increments and intervals as outlined in Table 3. Repeat the procedure above in the event of recurrence of Grade 3 symptoms. Permanently discontinue daratumumab upon the third occurrence of a Grade 3 or greater infusion reaction.
- Grade 4 (life threatening): Permanently discontinue daratumumab treatment.
- No dose reductions of daratumumab are recommended. Dose delay may be required to allow recovery of blood cell counts in the event of hematological toxicity.
- Daratumumab is for single use only. Prepare the solution for infusion using aseptic technique as follows:
- Calculate the dose (mg), total volume (mL) of daratumumab solution required and the number of daratumumab vials needed based on patient actual body weight.
- Check that the daratumumab solution is colorless to pale yellow. Do not use if opaque particles, discoloration or other foreign particles are present.
- Remove a volume of 0.9% Sodium Chloride Injection, USP from the infusion bag/container that is equal to the required volume of daratumumab solution.
- Withdraw the necessary amount of daratumumab solution and dilute to the appropriate volume by adding to the infusion bag/container containing 0.9% Sodium Chloride Injection, USP as specified in Table 3. Infusion bags/containers must be made of either polyvinyl chloride (PVC), polypropylene (PP), polyethylene (PE) or polyolefin blend (PP+PE). Dilute under appropriate aseptic conditions. Discard any unused portion left in the vial.
- Gently invert the bag/container to mix the solution. Do not shake.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The diluted solution may develop very small, translucent to white proteinaceous particles, as daratumumab is a protein. Do not use if visibly opaque particles, discoloration or foreign particles are observed.
- Since daratumumab does not contain a preservative, administer the diluted solution immediately at room temperature 15°C–25°C (59°F–77°F) and in room light. Diluted solution may be kept at room temperature for a maximum of 15 hours (including infusion time).
- If not used immediately, the diluted solution can be stored prior to administration for up to 24 hours at refrigerated conditions 2°C – 8°C (36°F–46°F) and protected from light. Do not freeze.
- Administer daratumumab as follows:
- If stored in the refrigerator, allow the solution to come to room temperature. Administer the diluted solution by intravenous infusion using an infusion set fitted with a flow regulator and with an in-line, sterile, non-pyrogenic, low protein-binding polyethersulfone (PES) filter (pore size 0.22 or 0.2 micrometer). Administration sets must be made of either polyurethane (PU), polybutadiene (PBD), PVC, PP or PE.
- Do not store any unused portion of the infusion solution for reuse. Any unused product or waste material should be disposed of in accordance with local requirements.
- Do not infuse daratumumab concomitantly in the same intravenous line with other agents.

Monitoring
Frequently monitor patients during the entire infusion. Interrupt daratumumab infusion for reactions of any severity and institute medical management as needed. Permanently discontinue daratumumab therapy for life-threatening (Grade 4) reactions. For patients with Grade 1, 2, or 3 reactions, reduce the infusion rate when re-starting the infusion.
IV Compatibility
There is limited information regarding the compatibility of Daratumumab and IV administrations.
Overdosage
The dose of daratumumab at which severe toxicity occurs is not known. In the event of an overdose, monitor patients for any signs or symptoms of adverse effects and provide appropriate supportive treatment.
Pharmacology

Mechanism of Action
CD38 is a transmembrane glycoprotein (48 kDa) expressed on the surface of hematopoietic cells, including multiple myeloma and other cell types and tissues and has multiple functions, such as receptor mediated adhesion, signaling, and modulation of cyclase and hydrolase activity. Daratumumab is an IgG1κ human monoclonal antibody (mAb) that binds to CD38 and inhibits the growth of CD38 expressing tumor cells by inducing apoptosis directly through Fc mediated cross linking as well as by immune-mediated tumor cell lysis through complement dependent cytotoxicity (CDC), antibody dependent cell mediated cytotoxicity (ADCC) and antibody dependent cellular phagocytosis (ADCP). A subset of myeloid derived suppressor cells (CD38+MDSCs), regulatory T cells (CD38+Tregs) and B cells (CD38+Bregs) are decreased by daratumumab.
Structure
There is limited information regarding Daratumumab Structure in the drug label.
Pharmacodynamics
NK cells express CD38 and are susceptible to daratumumab mediated cell lysis. Decreases in absolute counts and percentages of total NK cells (CD16+CD56+) and activated (CD16+CD56dim) NK cells in peripheral whole blood and bone marrow were observed with daratumumab treatment. Daratumumab as a large protein has a low likelihood of direct ion channel interactions. There is no evidence from non-clinical or clinical data to suggest that daratumumab has the potential to delay ventricular repolarization.
Pharmacokinetics
Over the dose range from 1 to 24 mg/kg as monotherapy or 1 to 16 mg/kg of daratumumab in combination with other treatments, increases in area under the concentration-time curve (AUC) were more than dose-proportional. Following the recommended dose of 16 mg/kg when daratumumab was administered as monotherapy or in combination therapy, the mean serum maximal concentration (Cmax) value at the end of weekly dosing, was approximately 2.7 to 3-fold higher compared to the mean serum Cmax following the first dose. The mean ± standard deviation (SD) trough serum concentration (Cmin) at the end of weekly dosing was 573 ± 332 µg/mL when daratumumab was administered as monotherapy and 502 ± 196 to 607 ± 231 µg/mL when daratumumab was administered as combination therapy. Daratumumab steady state was achieved approximately 5 months into the every 4-week dosing period (by the 21st infusion), and the mean ± SD ratio of Cmax at steady-state to Cmax after the first dose was 1.6 ± 0.5.
Distribution:
At the recommended dose of 16 mg/kg, the mean ± SD central volume of distribution was 4.7 ± 1.3 L when daratumumab was administered as monotherapy and 4.4 ± 1.5 L when daratumumab was administered as combination therapy.
Elimination:
Daratumumab clearance decreased with increasing dose and with multiple dosing. At the recommended dose of 16 mg/kg of daratumumab as monotherapy, the mean ± SD linear clearance was estimated to be 171.4 ± 95.3 mL/day. The mean ± SD estimated terminal half-life associated with linear clearance was 18 ± 9 days when daratumumab administered as monotherapy and 23 ± 12 days when daratumumab was administered as combination therapy.
Specific populations:
The following population characteristics have no clinically meaningful effect on the pharmacokinetics of daratumumab in patients administered daratumumab as monotherapy or as combination therapy: sex, age (31 to 84 years), mild [total bilirubin 1 to 1.5 times upper limit of normal (ULN) and any alanine transaminase (ALT)] and moderate (total bilirubin 1.5 to 3 times ULN and any ALT) hepatic impairment, or renal impairment [Creatinine clearance] (CLcr) 15 –89 mL/min]. The effect of severe (total bilirubin >3 times ULN and any ALT) hepatic impairment is unknown. Increasing body weight increased the central volume of distribution and clearance of daratumumab, supporting the body weight-based dosing regimen.
Drug interactions:
The coadministration of lenalidomide, pomalidomide or bortezomib with daratumumab did not affect the pharmacokinetics of daratumumab. The coadministration of daratumumab with bortezomib did not affect the pharmacokinetics of bortezomib.
Nonclinical Toxicology
No carcinogenicity or genotoxicity studies have been conducted with daratumumab. No animal studies have been performed to evaluate the potential effects of daratumumab on reproduction or development, or to determine potential effects on fertility in males or females.
Clinical Studies
Study 3, an open-label, randomized, active-controlled Phase 3 trial, compared treatment with daratumumab 16 mg/kg in combination with lenalidomide and low-dose dexamethasone (DRd) to treatment with lenalidomide and low-dose dexamethasone (Rd) in patients with multiple myeloma who had received at least one prior therapy. Lenalidomide (25 mg once daily orally on Days 1–21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/week (or a reduced dose of 20 mg/week for patients >75 years or body mass index [BMI] <18.5). On daratumumab infusion days, 20 mg of the dexamethasone dose was given as a pre-infusion medication and the remainder given the day after the infusion. For patients on a reduced dexamethasone dose, the entire 20 mg dose was given as a daratumumab pre-infusion medication. Dose adjustments for lenalidomide and dexamethasone were applied according to manufacturer's prescribing information. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 569 patients were randomized; 286 to the DRd arm and 283 to the Rd arm. The baseline demographic and disease characteristics were similar between the daratumumab and the control arm. The median patient age was 65 years (range 34 to 89 years), 11% were ≥75 years, 59% were male; 69% Caucasian, 18% Asian, and 3% African American. Patients had received a median of 1 prior line of therapy. Sixty-three percent (63%) of patients had received prior autologous stem cell transplantation (ASCT). The majority of patients (86%) received a prior PI, 55% of patients had received a prior immunomodulatory agent, including 18% of patients who had received prior lenalidomide; and 44% of patients had received both a prior PI and immunomodulatory agent. At baseline, 27% of patients were refractory to the last line of treatment. Eighteen percent (18%) of patients were refractory to a PI only, and 21% were refractory to bortezomib. Efficacy was evaluated by progression free survival (PFS) based on International Myeloma Working Group (IMWG) criteria.
Study 3 demonstrated an improvement in PFS in the DRd arm as compared to the Rd arm; the median PFS had not been reached in the DRd arm and was 18.4 months in the Rd arm (hazard ratio [HR]=0.37; 95% CI: 0.27, 0.52; p<0.0001), representing 63% reduction in the risk of disease progression or death in patients treated with DRd. Figure 1: Kaplan-Meier Curve of PFS in Study 3

Additional efficacy results from Study 3 are presented in Table 12 below.

In responders, the median time to response was 1 month (range: 0.9 to 13 months) in the DRd group and 1.1 months (range: 0.9 to 10 months) in the Rd group. The median duration of response had not been reached in the DRd group (range: 1+ to 19.8+ months) and was 17.4 months (range: 1.4 to 18.5+ months) in the Rd group.
With a median follow-up of 13.5 months, 75 deaths were observed; 30 in the DRd group and 45 in the Rd group.
Combination Treatment with Bortezomib and Dexamethasone
Study 4, an open-label, randomized, active-controlled Phase 3 trial, compared treatment with daratumumab 16 mg/kg in combination with bortezomib and dexamethasone (DVd), to treatment with bortezomib and dexamethasone (Vd). Bortezomib was administered by SC injection or IV infusion at a dose of 1.3 mg/m2 body surface area twice weekly for two weeks (Days 1, 4, 8, and 11) of repeated 21 day (3-week) treatment cycles, for a total of 8 cycles. Dexamethasone was administered orally at a dose of 20 mg on Days 1, 2, 4, 5, 8, 9, 11, and 12 of each of the 8 bortezomib cycles (80 mg/week for two out of three weeks of the bortezomib cycle) or a reduced dose of 20 mg/week for patients >75 years, BMI <18.5, poorly controlled diabetes mellitus or prior intolerance to steroid therapy. On the days of daratumumab infusion, 20 mg of the dexamethasone dose was administered as a pre-infusion medication. For patients on a reduced dexamethasone] dose, the entire 20 mg dose was given as a daratumumab pre-infusion medication. Bortezomib and dexamethasone were given for 8 three-week cycles in both treatment arms; whereas daratumumab was given until disease progression. However, dexamethasone 20 mg was continued as a daratumumab pre-infusion medication in the DVd arm. Dose adjustments for bortezomib and dexamethasone were applied according to manufacturer's prescribing information.
A total of 498 patients were randomized; 251 to the DVd arm and 247 to the Vd arm. The baseline demographic and disease characteristics were similar between the daratumumab and the control arm. The median patient age was 64 years (range 30 to 88 years); 12% were ≥75 years, 57% were male; 87% Caucasian, 5% Asian and 4% African American. Patients had received a median of 2 prior lines of therapy and 61% of patients had received prior autologous stem cell transplantation (ASCT). Sixty-nine percent (69%) of patients had received a prior PI (66% received bortezomib) and 76% of patients received an immunomodulatory agent (42% received lenalidomide). At baseline, 32% of patients were refractory to the last line of treatment and the proportions of patients refractory to any specific prior therapy were in general well balanced between the treatment groups. Thirty-three percent (33%) of patients were refractory to an immunomodulatory agent only, with 24% patients in the DVd arm and 33% of patients in the Vd arm respectively refractory to lenalidomide. Efficacy was evaluated by progression free survival (PFS) based on International Myeloma Working Group (IMWG) criteria.
Study 4 demonstrated an improvement in PFS in the DVd arm as compared to the Vd arm; the median PFS had not been reached in the DVd arm and was 7.2 months in the Vd arm (HR [95% CI]: 0.39 [0.28, 0.53]; p-value < 0.0001), representing a 61% reduction in the risk of disease progression or death for patients treated with DVd versus Vd. Figure 2: Kaplan-Meier Curve of PFS in Study 4

Additional efficacy results from Study 4 are presented in Table 13 below.

In responders, the median time to response was 0.8 months (range: 0.7 to 4 months) in the DVd group and 1.5 months (range: 0.7 to 5 months) in the Vd group. The median duration of response had not been reached in the DVd group (range: 1.4+ to 14.1+ months) and was 7.9 months (1.4+ to 12+ months) in the Vd group.
With a median follow-up of 7.4 months, 65 deaths were observed; 29 in the DVd group and 36 in the Vd group were observed.
Study 5 was an open-label trial in which 103 patients with multiple myeloma who had received a prior PI and an immunomodulatory agent, received 16 mg/kg daratumumab in combination with pomalidomide and low-dose dexamethasone until disease progression. Pomalidomide (4 mg once daily orally on Days 1-21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/ week (reduced dose of 20 mg/week for patients >75 years or body mass index [BMI] <18.5). On daratumumab infusion days, 20 mg of the dexamethasone dose was given as a pre-infusion medication and the remainder given the day after the infusion. For patients on a reduced dexamethasone dose, the entire 20 mg dose was given as a daratumumab pre-infusion medication.
The median patient age was 64 years (range: 35 to 86 years) with 8% of patients ≥75 years of age. Patients in the study had received a median of 4 prior lines of therapy. Seventy-four percent (74%) of patients had received prior ASCT. Ninety-eight percent (98%) of patients received prior bortezomib treatment, and 33% of patients received prior carfilzomib. All patients received prior lenalidomide treatment, with 98% of patients previously treated with the combination of bortezomib and lenalidomide. Eighty nine percent (89%) of patients were refractory to lenalidomide and 71% refractory to bortezomib; 64% of patients were refractory to bortezomib and lenalidomide.
Efficacy results were based on overall response rate as determined by Independent Review Committee using IMWG criteria (see table 14).

The median time to response was 1 month (range: 0.9 to 2.8 months). The median duration of response was 13.6 months (range: 0.9+ to 14.6+ months).
Study 1, was an open-label trial evaluating daratumumab monotherapy in patients with relapsed or refractory multiple myeloma who had received at least 3 prior lines of therapy including a proteasome inhibitor and an immunomodulatory agent or who were double-refractory to a proteasome inhibitor and an immunomodulatory agent. In 106 patients, daratumumab 16 mg/kg was administered with pre- and post-infusion medication. Treatment continued until unacceptable toxicity or disease progression.
The median patient age was 63.5 years (range: 31 to 84 years), 49% were male and 79% were Caucasian. Patients had received a median of 5 prior lines of therapy. Eighty percent of patients had received prior autologous stem cell transplantation (ASCT). Prior therapies included bortezomib (99%), lenalidomide (99%), pomalidomide (63%) and carfilzomib (50%). At baseline, 97% of patients were refractory to the last line of treatment, 95% were refractory to both, a proteasome inhibitor (PI) and immunomodulatory agent, and 77% were refractory to alkylating agents.
Efficacy results were based on overall response rate as determined by the Independent Review Committee assessment using IMWG criteria (see table 15).

The median time to response was 1 month (range: 0.9 to 5.6 months). The median duration of response was 7.4 months (range: 1.2 to 13.1+ months).
Study 2 was an open-label dose escalation trial evaluating daratumumab monotherapy in patients with relapsed or refractory multiple myeloma who had received at least 2 different cytoreductive therapies. In 42 patients, daratumumab 16 mg/kg was administered with pre- and post-infusion medication. Treatment continued until unacceptable toxicity or disease progression.
The median patient age was 64 years (range: 44 to 76 years), 64% were male and 76% were Caucasian. Patients in the study had received a median of 4 prior lines of therapy. Seventy-four percent of patients had received prior ASCT. Prior therapies included bortezomib (100%), lenalidomide (95%), pomalidomide (36%) and carfilzomib (19%). At baseline, 76% of patients were refractory to the last line of treatment, 64% of patients were refractory to both, a PI and an immunomodulatory agent, and 60% of patients were refractory to alkylating agents.
Overall response rate was 36% (95% CI: 21.6, 52.0%) with 1 CR and 3 VGPR. The median time to response was 1 month (range: 0.5 to 3.2 months). The median duration of response was not estimable (range: 2.2 to 13.1+ months).
How Supplied
Daratumumab is a colorless to pale yellow, preservative-free solution for intravenous infusion supplied as:
- NDC 57894-502-05 contains one 100 mg/5 mL single-dose vial
- NDC 57894-502-20 contains one 400 mg/20 mL single-dose vial
Storage
Store in a refrigerator at 2ºC to 8ºC (36ºF to 46ºF). Do not freeze or shake. Protect from light. This product contains no preservative.
Images
Drug Images
Package and Label Display Panel


Patient Counseling Information
- Infusion Reactions
- Advise patients to seek immediate medical attention for any of the following signs and symptoms of infusion reactions: itchy, runny or blocked nose; chills, nausea, throat irritation, cough, headache, shortness of breath or difficulty breathing
- Neutropenia
- Advise patients that if they have a fever, they should contact their healthcare professional.
- Thrombocytopenia
- Interference with laboratory tests
- Advise patients to inform healthcare providers including blood transfusion centers/personnel that they are taking daratumumab, in the event of a planned transfusion.
- Advise patients that daratumumab can affect the results of some tests used to determine complete response in some patients and additional tests may be needed to evaluate response.
Precautions with Alcohol
Alcohol-Daratumumab interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
Darzalex
Look-Alike Drug Names
There is limited information regarding daratumumab Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.