Incidentaloma pathophysiology
|
Incidentaloma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Incidentaloma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Incidentaloma pathophysiology |
|
Risk calculators and risk factors for Incidentaloma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Mohammed Abdelwahed M.D[2]
Overview
The pathophysiology of adrenal incidentaloma depends on nature of the mass and its function. Incidentalomas are adrenal tumors that often discovered as an incidental finding. Malignancy is an uncommon cause of adrenal incidentaloma in patients without a known diagnosis of cancer. Incidentalomas may secrete cortisol. Cushing's syndrome is linked to hypercortisolism which can develop by excess ACTH secretion or excess cortisol secretion by adrenal glands. Incidentalomas also may secrete catecholamines and in this case it is considered pheochromocytoma. Pheochromocytoma arises from chromaffin cells of the adrenal medulla and sympathetic ganglia. Malignant and benign pheochromocytomas share the same biochemical and histological features, the only difference is to have a distant spread or be locally invasive. It may be sporadic, but some occur as a component of hereditary cancer syndromes such as Li-Fraumeni syndrome, Beckwith-Wiedemann syndrome, and Multiple endocrine neoplasia type 1. Genetic base of sporadic incidentaloma is mutations in TP53 gene, located on chromosome 17p13. A role for the TP53 tumor suppressor gene in sporadic adrenocortical carcinoma. On gross pathology, adrenocortical adenoma is a yellow, well circumscribed tumor in the adrenal cortex, which is usually 2–5 cm in diameter. The color of tumor, as with adrenal cortex as a whole, is due to the stored lipid (mainly cholesterol), from which the cortical hormones are synthesized.
Pathophysiology
Incidentalomas are adrenal tumors that are often discovered as an incidental finding.[1][2][3]
- Most incidentalomas are nonfunctional, 9% are found to secrete low levels of cortisol, 4% are pheochromocytomas, and 1.5% are aldosteronomas.
- Malignancy is an uncommon cause of adrenal incidentaloma in patients without a known diagnosis of cancer.
- Frequency of primary adrenal carcinoma is approximately 2 to 5 percent; and 0.7 to 2.5% have non-adrenal metastases to the adrenal gland.
- Most adrenocortical carcinomas are sporadic, but some occur as a component of hereditary cancer syndromes.
Subclinical Cushing's syndrome pathogenesis
The pathophysiology of Cushing's syndrome is linked to hypercortisolism which can develop by excess ACTH secretion or excess cortisol secretion by adrenal glands. The underlying mechanisms are usually genetic mutations or overexpression of proteins:[4]
- Benign adrenocortical adenoma: Common defects leading to adrenocortical adenoma are mutations or activation of the cAMP-dependent or β-catenin signaling pathways and aberrant expression and function of various G-protein-coupled receptors (GPCR).[5]
- Adrenal cortical carcinoma: It is associated with germline TP53 mutations and MEN syndrome.[6]
- Bilateral adrenal hyperplasia: It is associated with MEN1, familial adenomatous polyposis, and fumarate hydratase gene mutations. Several inactivating mutations of armadillo repeat containing 5 genes are also identified.
Mechanism of cortisol secretion
The secretion of cortisol is controlled by hypothalamic-pituitary axis by the following mechanism:[7]
- Paraventricular nuclei in the hypothalamus release corticotropin releasing hormone (CRH).
- CRH is transferred to anterior pituitary via the portal veins.
- CRH stimulates the activity of corticotrophs; cells that produce proopiomelanocortin (POMC) in the anterior pituitary.
- Corticotrophs produce adrenocorticotropic hormone (ACTH) by the post-translational modification of POMC.
- ACTH is drained into systemic circulation via the pituitary capillaries and stimulates the adrenal cortex (zona fasciculata) to produce cortisol.
- Cortisol acts on hypothalamus and pituitary through a feedback mechanism to regulate the secretion of CRH and ACTH.
Pathogenesis of pheochromocytoma
Pheochromocytoma arises from chromaffin cells of the adrenal medulla and sympathetic ganglia. Malignant and benign pheochromocytomas share the same biochemical and histological features, the only difference is to have a distant spread or be locally invasive.
Basic physiology of catecholamines
Catecholamines act on nearly all body tissues. Its actions vary by tissue type and tissue expression of adrenergic receptors.[8]
- Epinephrine is a non-selective agonist of all adrenergic receptors, including the major sub-types α1, α2, β1, β2, and β3:
- Binding to α1 receptors causes vasoconstriction. Blood vessels with α1-adrenergic receptors are present in the skin, the sphincters of the gastrointestinal system, kidney (renal artery) and brain. During the fight-or-flight response vasoconstriction results in decreased blood flow to these organs.
- Binding to α2 receptors inhibits insulin secretion by the pancreas, stimulates glycogenolysis in the liver and muscle, and stimulates glycolysis and inhibits insulin-mediated glycogenesis in muscle. It suppresses the release of norepinephrine by negative feedback.
- Binding to β2 receptors causes smooth muscle relaxation in the uterus, GI tract, urinary detrusor muscle of bladder wall, and bronchi. It also causes dilatation of smaller coronary arteries, hepatic artery, skeletal muscle arteries.
- Binding to β1 receptors causes renin release from juxtaglomerular cells and lipolysis in adipose tissue. It Increases cardiac output via below mechanisms:
- Increase in heart rate in sinoatrial node.
- Increase in atrial cardiac muscle contractility.
- Increases in contractility and automaticity of ventricular cardiac muscle.
- Increases in conduction and automaticity of atrioventricular node.
Genetics
Sporadic cases genetics
- TP53 gene, located on chromosome 17p13, is the most frequently mutated gene in human cancers. A role for the TP53 tumor suppressor gene in sporadic ACCs is suggested by the frequent finding of loss of heterozygosity (LOH) at the 17p13 locus in sporadic ACCs.[9]
- Although loss of heterozygosity at 17p13 is common, only approximately one-third of these tumors have a mutation of TP53. This suggests that another as yet unidentified suppressor gene is present in this locus.[10]
- Another chromosomal locus that is strongly implicated in the pathogenesis of ACC is 11p, the area of abnormality in Beckwith-Wiedemann syndrome and the site of the insulin-like growth factor-2 (IGF-2) gene. LOH at the 11p15 locus and overexpression of IGF-2 have been associated with the malignant phenotype in sporadic ACCs.[11] However, other growth-related tumor suppressor genes at this locus may also be involved.[12]
Most adrenocortical tumors are monoclonal, suggesting that they result from accumulated genetic abnormalities, such as activation of proto-oncogenes and inactivation of tumor suppressor genes.
- Beta-catenin mutations (CTNNB1):
- Constitutive activation of beta-catenin in the Wnt signaling pathway has been identified as a frequent alteration in benign and malignant adrenocortical tumors[13].
- The increased occurrence of adrenal tumors in patients with mutations of adenomatous polyposis coli (APC) suggested that the Wnt/beta-catenin pathway could be involved in adrenal tumorigenesis.[14]
- This pathway is essential for embryonic development of the adrenal, and its ectopic constitutive activation is associated with cancer development in a number of tissues.[15]
- Aberrant receptors:
- Somatic mutations of protein kinase A (PKA) catalytic subunit (PRKACA) were identified in patients with overt Cushing's syndrome but not in adenomas secreting less cortisol.[16]
- In additional reports, the same mutation was found in over 50 percent of patients with Cushing's syndrome due to adrenal adenomas.[17]
- The most frequent hotspot p.Leu206Arg mutation is located in the active cleft of the catalytic subunit, inactivating the site where the regulatory subunit RII-beta usually binds, thus causing a constitutive PKA activation.
Mutations in aldosterone-producing adenomas
- The most frequent causes of primary aldosteronism include bilateral idiopathic hyperplasia and unilateral aldosterone-producing adenoma.[18]
- Somatic mutations in KCNJ5 have been identified in patients with primary aldosteronism due to APAs.
- These mutations are more common in women than men; APAs with KCNJ5 mutations are larger than those without mutations.
- Somatic mutations in other important genes implicated in regulation of aldosterone synthesis (ATP1A1, ATP2B3, CACNA1D, CTNNB1, ARMC5) have also been identified.
Associated Conditions
- Most adrenocortical carcinomas are sporadic, but some occur as a component of hereditary cancer syndromes.[19][20]
- Hereditary cancer syndromes:
- Li-Fraumeni syndrome (associated with inactivating mutations of the TP53 tumor suppressor gene on chromosome 17p):
- Breast cancer
- Soft tissue and bone sarcoma
- Brain tumors
- Beckwith-Wiedemann syndrome (associated with abnormalities in 11p15):
- Multiple endocrine neoplasia type 1 (MEN1) (associated with inactivating mutations of the MEN1 gene on chromosome 11q):
- Li-Fraumeni syndrome (associated with inactivating mutations of the TP53 tumor suppressor gene on chromosome 17p):
Gross Pathology
- On gross pathology, adrenocortical adenoma is a solitary unilateral well-circumscribed mass.
- Size usually varies from 2–5 cm in diameter.
- The color of tumor is yellow due to the stored lipid (cholesterol) from which the cortical hormones are synthesized.
- Weight usually < 50 grams.
- Functional ACA may result in atrophy of ipsilateral or contralateral adrenal cortex.
- On gross pathology, adrenocortical carcinomas are often large ( > 5 cm in largest diameter).
- Cut surface is full of areas of hemorrhage and necrosis.
- ACC color ranges from brown to orange depending on the lipid content of their cells.
- ACC is unencapsulated, often soft and friable.
- On gross pathology, pheochromocytoma varies from small to large and usually associated with hemorrhage and necrosis.[21]
- Pheochromocytoma are usually lobulated and small tumors have compressed adrenal gland.
- Familial tumors are bilateral.
- It may be associated with hyperplasia in the adjacent medulla.
- Chromaffin reaction: Fresh tumor cut section turns dark brown if add potassium dichromate at pH 5-6.
- On gross pathology, myelolipoma usually occurs unilateral (one adrenal gland).
- Myelolipoma diameter ranges from small (few millimeters) to large (34 centimeters).
- The cut surface has colors varying from yellow to red to brown, depending on the distribution of fat and blood.
-
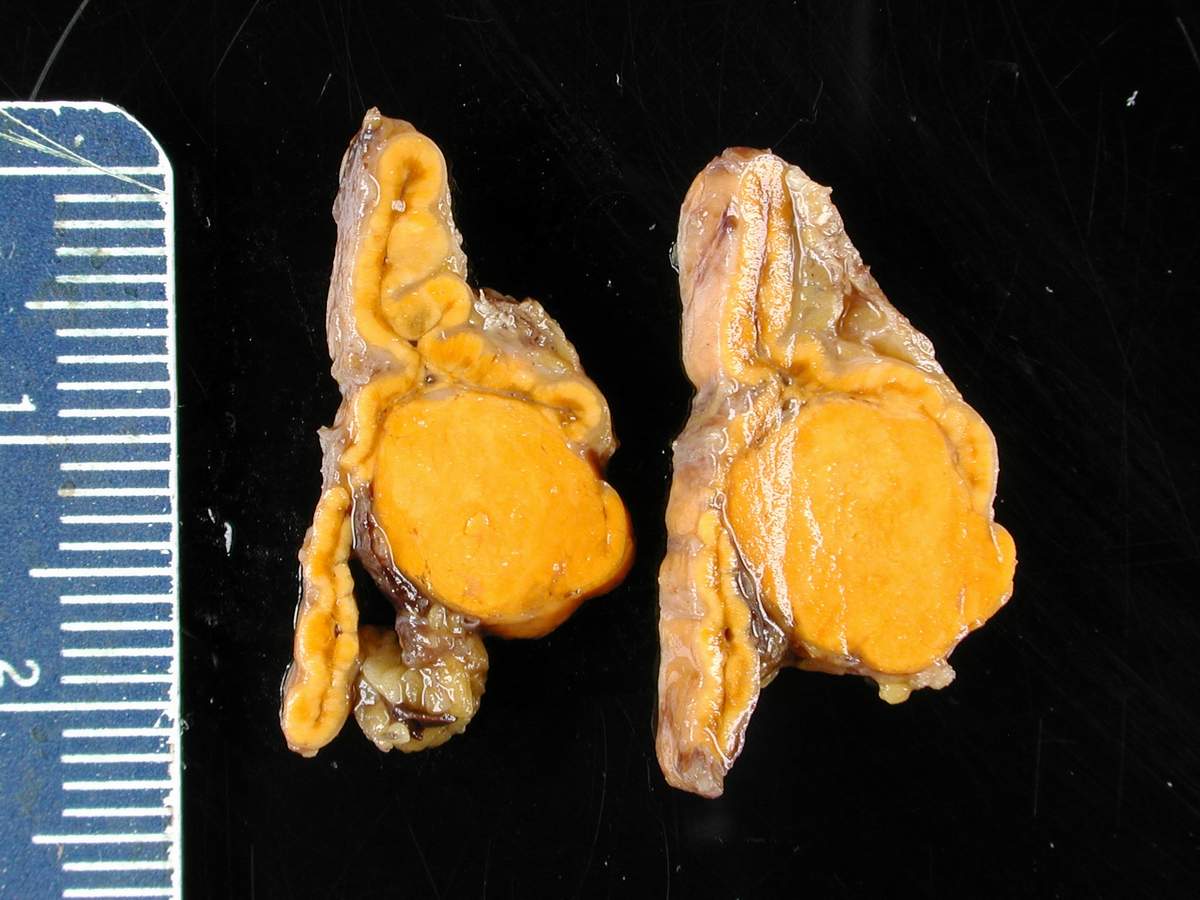 Adrenocortical adenoma gross pathology, source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524
Adrenocortical adenoma gross pathology, source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524 -
![Adrenocortical carcinoma gross pathology, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719487](/images/1/18/Adrenal_cortical_carcinoma.JPG) Adrenocortical carcinoma gross pathology, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719487
Adrenocortical carcinoma gross pathology, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719487 -
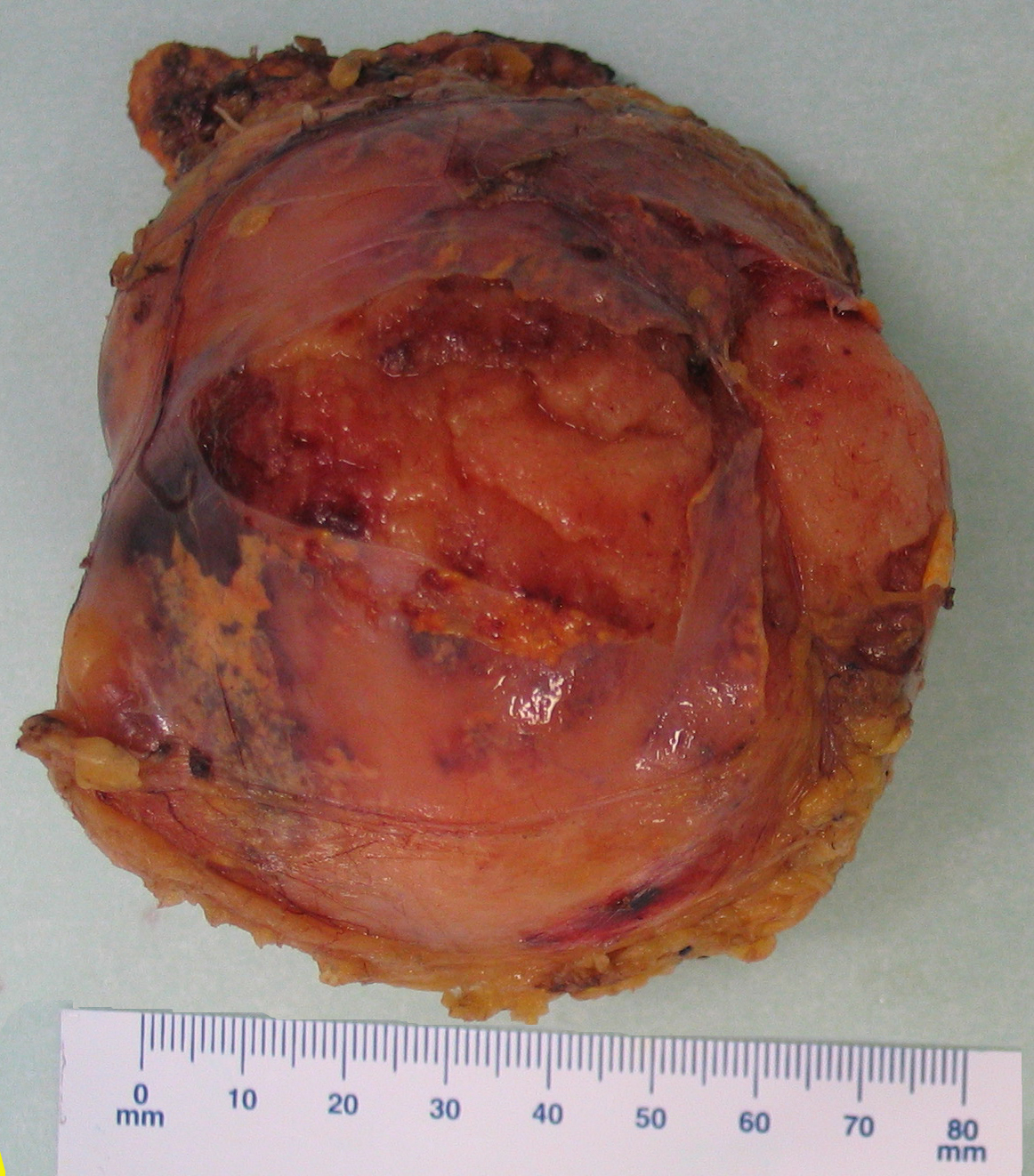 Myelolipoma gross pathology,source: By Mattopaedia - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5668284
Myelolipoma gross pathology,source: By Mattopaedia - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5668284 -
![Bilateral pheochromocytoma in MEN2. Gross image, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=4288117](/images/5/5f/Bilateral_pheo_MEN2.jpg) Bilateral pheochromocytoma in MEN2. Gross image, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=4288117
Bilateral pheochromocytoma in MEN2. Gross image, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=4288117

![Adrenocortical carcinoma gross pathology, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719487](/index.php/File:Adrenal_cortical_carcinoma.JPG)

![Bilateral pheochromocytoma in MEN2. Gross image, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=4288117](/index.php/File:Bilateral_pheo_MEN2.jpg)
Microscopic Pathology
Adrenal adenoma and carcinoma
- On microscopic examination, the tumor usually displays sheets of atypical cells with some resemblance to the cells of the normal adrenal cortex. The presence of invasion and mitotic activity help differentiate small cancers from adrenocortical adenomas.[5][22]
- In comparison to surrounding adrenal gland, Adrenal cortical adenoma cells are larger, different cytoplasm, increased variation in nuclear size
- Distinct cell borders, cells have abundant foamy cytoplasm reminiscent of zona fasciculata
- Balloon cells: clusters of cells with enlarged lipid-rich cytoplasm
Pheochromocytoma
On microscopic pathology, pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma.[23][24]
Myelolipoma
- Islands of hematopoietic cells and mature fat.
- Usually show normal trilineage hematopoiesis, but often with markedly increased megakaryocytes.
- Larger tumors may have hemorrhage, necrosis, calcification and cysts.
- Rarely may have areas of fibromyxoid degeneration resembling low grade fibromyxoid sarcoma.
-
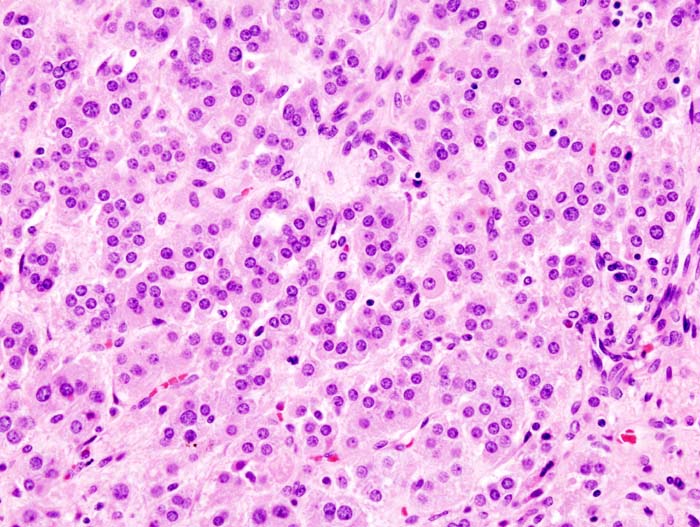 Adrenal adenoma microscopic picture, source: By Michael Feldman, MD, PhDUniversity of Pennsylvania School of Medicine - CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535950
Adrenal adenoma microscopic picture, source: By Michael Feldman, MD, PhDUniversity of Pennsylvania School of Medicine - CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535950 -
![Adrenocortical carcinoma microscopic picture, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719510](/images/a/a0/Adrenal_cortical_carcinoma_%281%29.jpg) Adrenocortical carcinoma microscopic picture, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719510
Adrenocortical carcinoma microscopic picture, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719510 -
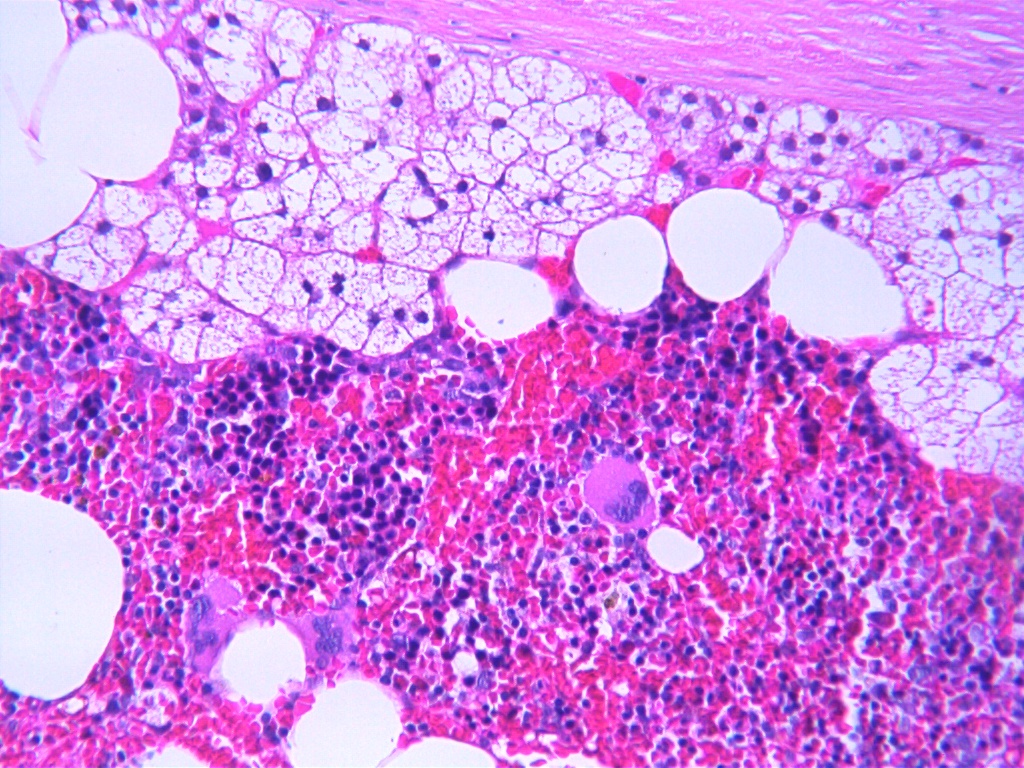 Myelolipoma microscopic picture, source: By Mattopaedia - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5688142
Myelolipoma microscopic picture, source: By Mattopaedia - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5688142 -
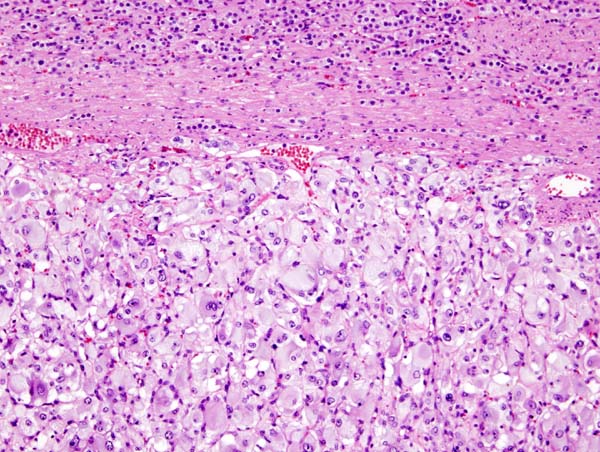 Micrograph of pheochromocytoma, source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524
Micrograph of pheochromocytoma, source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524 -
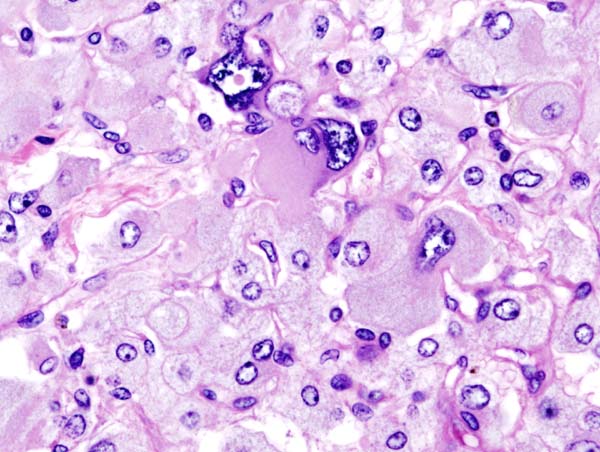 Histopathology of adrenal pheochromocytoma. Adrenectomy specimen, source: Wikipedia
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen, source: Wikipedia -
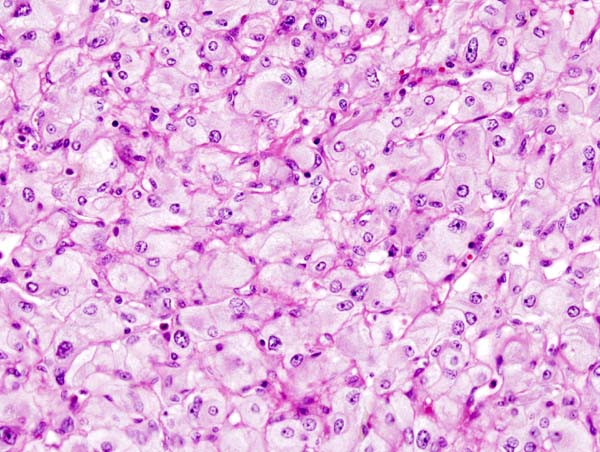 Micrograph of pheochromocytoma, source: Wikipedia
Micrograph of pheochromocytoma, source: Wikipedia -
Micrograph of pheochromocytoma, source: Wikipedia

![Adrenocortical carcinoma microscopic picture, source: By AFIP Atlas of Tumor Pathology - [1], Public Domain, https://commons.wikimedia.org/w/index.php?curid=6719510](/index.php/File:Adrenal_cortical_carcinoma_(1).jpg)

_histopathology.jpg)
_histopathology.jpg)
_histopathology.jpg)
References
- ↑ Grumbach MM, Biller BM, Braunstein GD, Campbell KK, Carney JA, Godley PA; et al. (2003). "Management of the clinically inapparent adrenal mass ("incidentaloma")". Ann Intern Med. 138 (5): 424–9. PMID 12614096.
- ↑ Young WF (2000). "Management approaches to adrenal incidentalomas. A view from Rochester, Minnesota". Endocrinol Metab Clin North Am. 29 (1): 159–85, x. PMID 10732270.
- ↑ Sidhu S, Sywak M, Robinson B, Delbridge L (2004). "Adrenocortical cancer: recent clinical and molecular advances". Curr Opin Oncol. 16 (1): 13–8. PMID 14685087.
- ↑ Lacroix A, Feelders RA, Stratakis CA, Nieman LK (2015). "Cushing's syndrome". Lancet. 386 (9996): 913–27. doi:10.1016/S0140-6736(14)61375-1. PMID 26004339.
- ↑ Raff H, Carroll T (2015). "Cushing's syndrome: from physiological principles to diagnosis and clinical care". J Physiol. 593 (3): 493–506. doi:10.1113/jphysiol.2014.282871. PMC 4324701. PMID 25480800.
- ↑ Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM; et al. (2014). "Adrenocortical carcinoma". Endocr Rev. 35 (2): 282–326. doi:10.1210/er.2013-1029. PMC 3963263. PMID 24423978.
- ↑ Raff H, Carroll T (2015). "Cushing's syndrome: from physiological principles to diagnosis and clinical care". J Physiol. 593 (3): 493–506. doi:10.1113/jphysiol.2014.282871. PMC 4324701. PMID 25480800.
- ↑ Arnall DA, Marker JC, Conlee RK, Winder WW (1986). "Effect of infusing epinephrine on liver and muscle glycogenolysis during exercise in rats". Am J Physiol. 250 (6 Pt 1): E641–9. PMID 3521311.
- ↑ Gicquel C, Bertagna X, Gaston V, Coste J, Louvel A, Baudin E; et al. (2001). "Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors". Cancer Res. 61 (18): 6762–7. PMID 11559548.
- ↑ Libè R, Groussin L, Tissier F, Elie C, René-Corail F, Fratticci A; et al. (2007). "Somatic TP53 mutations are relatively rare among adrenocortical cancers with the frequent 17p13 loss of heterozygosity". Clin Cancer Res. 13 (3): 844–50. doi:10.1158/1078-0432.CCR-06-2085. PMID 17289876.
- ↑ Gicquel C, Raffin-Sanson ML, Gaston V, Bertagna X, Plouin PF, Schlumberger M; et al. (1997). "Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: study on a series of 82 tumors". J Clin Endocrinol Metab. 82 (8): 2559–65. doi:10.1210/jcem.82.8.4170. PMID 9253334.
- ↑ Bourcigaux N, Gaston V, Logié A, Bertagna X, Le Bouc Y, Gicquel C (2000). "High expression of cyclin E and G1 CDK and loss of function of p57KIP2 are involved in proliferation of malignant sporadic adrenocortical tumors". J Clin Endocrinol Metab. 85 (1): 322–30. doi:10.1210/jcem.85.1.6303. PMID 10634406.
- ↑ Mazzuco TL, Durand J, Chapman A, Crespigio J, Bourdeau I (2012). "Genetic aspects of adrenocortical tumours and hyperplasias". Clin Endocrinol (Oxf). 77 (1): 1–10. doi:10.1111/j.1365-2265.2012.04403.x. PMID 22471738.
- ↑ Smith TG, Clark SK, Katz DE, Reznek RH, Phillips RK (2000). "Adrenal masses are associated with familial adenomatous polyposis". Dis Colon Rectum. 43 (12): 1739–42. PMID 11156460.
- ↑ Kikuchi A (2003). "Tumor formation by genetic mutations in the components of the Wnt signaling pathway". Cancer Sci. 94 (3): 225–9. PMID 12824913.
- ↑ Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A; et al. (2014). "Constitutive activation of PKA catalytic subunit in adrenal Cushing's syndrome". N Engl J Med. 370 (11): 1019–28. doi:10.1056/NEJMoa1310359. PMC 4727447. PMID 24571724.
- ↑ Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assié G, Weigand I; et al. (2016). "Genetic Landscape of Sporadic Unilateral Adrenocortical Adenomas Without PRKACA p.Leu206Arg Mutation". J Clin Endocrinol Metab. 101 (9): 3526–38. doi:10.1210/jc.2016-1586. PMID 27389594.
- ↑ Monticone S, Castellano I, Versace K, Lucatello B, Veglio F, Gomez-Sanchez CE; et al. (2015). "Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas". Mol Cell Endocrinol. 411: 146–54. doi:10.1016/j.mce.2015.04.022. PMC 4474471. PMID 25958045.
- ↑ Koch CA, Pacak K, Chrousos GP (2002). "The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors". J Clin Endocrinol Metab. 87 (12): 5367–84. doi:10.1210/jc.2002-021069. PMID 12466322.
- ↑ Lynch HT, Radford B, Lynch JF (1990). "SBLA syndrome revisited". Oncology. 47 (1): 75–9. PMID 2300390.
- ↑ Sajjanar AB, Athanikar VS, Dinesh US, Nanjappa B, Patil PB (2015). "Non Functional Unilateral Adrenal Myelolipoma, A Case Report". J Clin Diagn Res. 9 (6): ED03–4. doi:10.7860/JCDR/2015/13209.6070. PMC 4525519. PMID 26266130.
- ↑ Mondal SK, Dasgupta S, Jain P, Mandal PK, Sinha SK (2013). "Histopathological study of adrenocortical carcinoma with special reference to the Weiss system and TNM staging and the role of immunohistochemistry to differentiate it from renal cell carcinoma". J Cancer Res Ther. 9 (3): 436–41. doi:10.4103/0973-1482.119329. PMID 24125979.
- ↑ Bezuglova TV (2003). "Criteria for predicting the outcome of pheochromocytoma by the immunohistochemical and electron microscopic findings". Bull Exp Biol Med. 136 (4): 408–10. PMID 14714096.
- ↑ Sporny S, Musiał J (2005). "[Markers of malignancy in pheochromocytomas]". Endokrynol Pol. 56 (6): 946–51. PMID 16821216.