Pasireotide
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Vignesh Ponnusamy, M.B.B.S. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Pasireotide is a somatostatin analog that is FDA approved for the treatment of Cushing’s disease for whom pituitary surgery is not an option or has not been curative. Common adverse reactions include diarrhea, nausea, hyperglycemia, cholelithiasis, headache, abdominal pain, fatigue, and diabetes mellitus.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Cushing’s Disease
- SIGNIFOR is indicated for the treatment of adult patients with Cushing’s disease for whom pituitary surgery is not an option or has not been curative.
- The recommended dosage range of SIGNIFOR is 0.3 to 0.9 mg by subcutaneous injection twice a day. The recommended initial dose is either 0.6 mg or 0.9 mg twice a day. Titrate dose based on response and tolerability.
- Patients should be evaluated for a treatment response [clinically meaningful reduction in 24-hour urinary free cortisol (UFC) levels and/or improvement in signs or symptoms of the disease] and should continue receiving therapy with SIGNIFOR as long as benefit is derived. Maximum urinary free cortisol reduction is typically seen by two months of treatment. For patients who are started on 0.6 mg twice a day, a dosage increase to 0.9 mg twice a day may be considered based on the response to the treatment, as long as the 0.6 mg dosage is well tolerated by the patient.
- Management of suspected adverse reactions may require temporary dose reduction of SIGNIFOR. Dose reduction by 0.3 mg decrements per injection is suggested.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Pasireotide in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Pasireotide in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Pasireotide in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Pasireotide in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Pasireotide in pediatric patients.
Contraindications
- None.
Warnings
Precautions
- Hypocortisolism
- Treatment with SIGNIFOR leads to suppression of adrenocorticotropic hormone (ACTH) secretion in Cushing’s disease. Suppression of ACTH may lead to a decrease in circulating levels of cortisol and potentially hypocortisolism.
- Monitor and instruct patients on the signs and symptoms associated with hypocortisolism (e.g. weakness, fatigue, anorexia, nausea, vomiting, hypotension, hyponatremia or hypoglycemia). If hypocortisolism occurs, consider temporary dose reduction or interruption of treatment with SIGNIFOR, as well as temporary, exogenous glucocorticoid replacement therapy.
- Hyperglycemia and Diabetes
- Elevations in blood glucose levels have been seen in healthy volunteers and patients treated with SIGNIFOR. In the Phase III trial, the development of pre-diabetes and diabetes was observed. In this trial, nearly all patients—including those with normal glucose status at baseline, pre-diabetes, and diabetes—developed worsening glycemia in the first two weeks of treatment. Cushing’s disease patients with poor glycemic control (as defined by HbA1c values >8% while receiving anti-diabetic therapy) may be at a higher risk of developing severe hyperglycemia and associated complications, e.g. ketoacidosis.
- Because of this predictable adverse reaction, the glycemic status [[[fasting plasma glucose]] (FPG) or hemoglobin A1c (HbA1c)] should be assessed prior to starting treatment with SIGNIFOR. In patients with uncontrolled diabetes mellitus intensive anti-diabetic therapy should be initiated prior to treatment with SIGNIFOR. Self-monitoring of blood glucose and/or FPG assessments should be done every week for the first two to three months and periodically thereafter, as clinically appropriate. After treatment discontinuation, glycemic monitoring (e.g. FPG or HbA1c) should be done according to clinical practice. Patients who were initiated on anti-diabetic therapy as a result of SIGNIFOR may require closer monitoring after discontinuation of SIGNIFOR, especially if the anti-diabetic therapy has a risk of causing hypoglycemia.
- If hyperglycemia develops in a patient treated with SIGNIFOR, the initiation or adjustment of anti-diabetic treatment is recommended. The optimal treatment for the management of SIGNIFOR-induced hyperglycemia is not known. If uncontrolled hyperglycemia persists, despite appropriate medical management, the dose of SIGNIFOR should be reduced or discontinued.
- Bradycardia and QT Prolongation
- Bradycardia
- Bradycardia has been reported with the use of SIGNIFOR. Patients with cardiac disease and/or risk factors for bradycardia, such as history of clinically significant bradycardia, high-grade heart block, or concomitant use of drugs associated with bradycardia, should be carefully monitored. Dose adjustments of beta-blockers, calcium channel blockers, or correction of electrolyte disturbances may be necessary.
- QT Prolongation
- SIGNIFOR is associated with QT prolongation. In two thorough QT studies with SIGNIFOR, QT prolongation occurred at therapeutic and supra-therapeutic doses. SIGNIFOR should be used with caution in patients who are at significant risk of developing prolongation of QTc, such as those:
- with congenital long QT prolongation.
- with uncontrolled or significant cardiac disease including recent myocardial infarction, congestive heart failure, unstable angina or clinically significant bradycardia.
- on anti-arrhythmic therapy or other substances that are known to lead to QT prolongation.
- with hypokalemia and/or hypomagnesemia.
- A baseline ECG is recommended prior to initiating therapy with SIGNIFOR and monitoring for an effect on the QTc interval is advisable. Hypokalemia and hypomagnesemia must be corrected prior to SIGNIFOR administration and should be monitored periodically during therapy.
- Liver Test Elevations
- In the Phase III trial, 5% of patients had an ALT or AST level greater than 3 times the upper limit of normal (ULN). In the entire clinical development program of SIGNIFOR, there were 4 cases of concurrent elevations in ALT (alanine aminotransferase) greater than 3 x ULN and bilirubin greater than 2 x ULN: one patient with Cushing’s disease and three healthy volunteers. In these cases, total bilirubin elevations were seen either concomitantly or preceding the transaminase elevation.
- Monitoring of liver tests should be done after 1 to 2 weeks on treatment, then monthly for 3 months, and every 6 months thereafter. If ALT is normal at baseline and elevations of ALT of 3-5 times the ULN are observed on treatment, repeat the test within a week or within 48 hours if exceeding 5 times ULN. If ALT is abnormal at baseline and elevations of ALT of 3-5 times the baseline values are observed on treatment, repeat the test within a week or sooner if exceeding 5 times ULN. Tests should be done in a laboratory that can provide same-day results. If the values are confirmed or rising, interrupt SIGNIFOR treatment and investigate for probable cause of the findings, which may or may not be SIGNIFOR-related. Serial measures of ALT, aspartate aminotransferase, alkaline phosphatase, and total bilirubin, should be done weekly, or more frequently, if any value exceeds 5 times the baseline value in case of abnormal baselines or 5 times the ULN in case of normal baselines. If resolution of abnormalities to normal or near normal occurs, resuming treatment with SIGNIFOR may be done cautiously, with close observation, and only if some other likely cause has been found.
- Cholelithiasis
- Cholelithiasis has been frequently reported in clinical studies with SIGNIFOR. Ultrasonic examination of the gallbladder before, and at 6- to 12-month intervals during SIGNIFOR therapy is recommended.
- Monitoring for Deficiency of Pituitary Hormones
- As the pharmacological activity of SIGNIFOR mimics that of somatostatin, inhibition of pituitary hormones, other than ACTH, may occur. Monitoring of pituitary function (e.g., TSH/free T4, GH/IGF-1) should occur prior to initiation of therapy with SIGNIFOR and periodically during treatment should be considered as clinically appropriate. Patients who have undergone transsphenoidal surgery and pituitary irradiation are particularly at increased risk for deficiency of pituitary hormones.
Adverse Reactions
Clinical Trials Experience
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
- A total of 162 Cushing’s disease patients were exposed to SIGNIFOR in the Phase III study. At study entry, patients were randomized to receive twice a day (b.i.d.) doses of either 0.6 mg or 0.9 mg of SIGNIFOR given subcutaneously. The mean age of patients was approximately 40 years old with a predominance of female patients (78%). The majority of the patients had persistent or recurrent Cushing’s disease (83%) and few patients (≤ 5%) in either treatment group had received previous pituitary irradiation. The median exposure to the treatment was 10.4 months (0.03-37.8) with 68% of patients having at least six-months exposure.
- In the Phase III trial, adverse reactions were reported in 98% of patients. The most common adverse reactions (frequency ≥20% in either group) were diarrhea, nausea, hyperglycemia, cholelithiasis, headache, abdominal pain, fatigue, and diabetes mellitus. There were no deaths during the study. Serious adverse events were reported in 25% of patients. Adverse events leading to study discontinuation were reported in 17% of patients.
- Adverse reactions with an overall frequency higher than 5% are presented in Table 1 by randomized dose group and overall. Adverse reactions are ranked by frequency, with the most frequent reactions listed first.
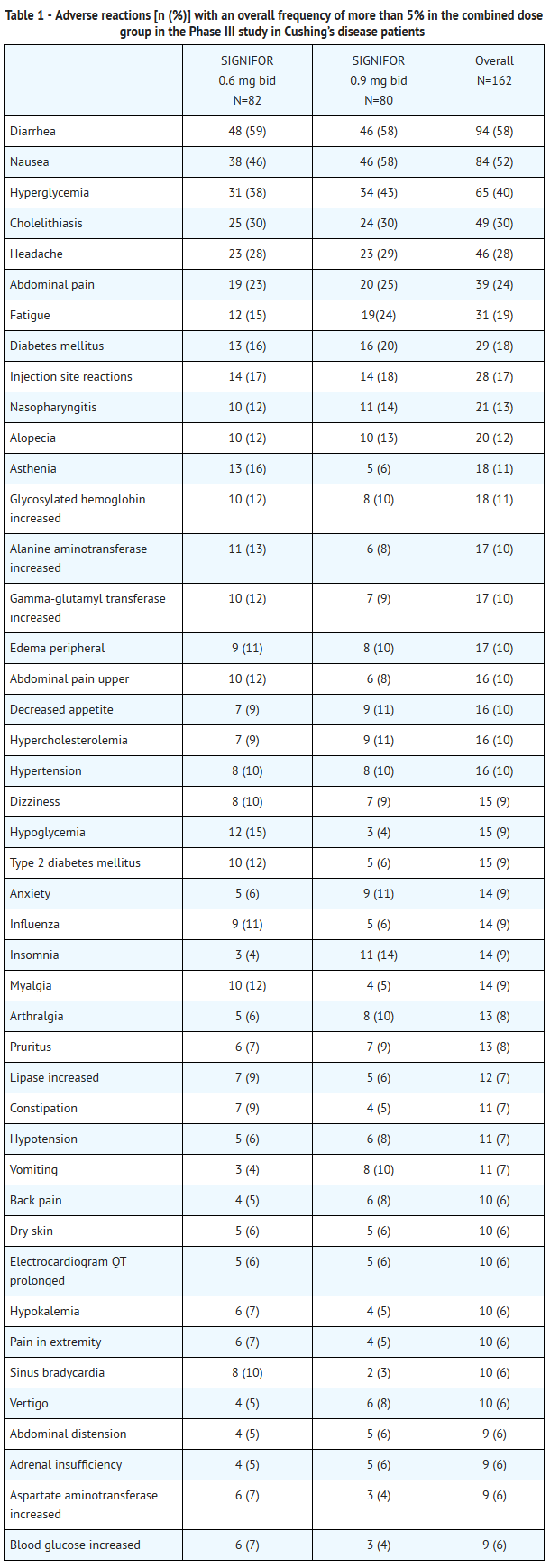
This image is provided by the National Library of Medicine.

- Other notable adverse reactions which occurred with a frequency less than 5% were: anemia (4%); blood amylase increased (2%) and prothrombin time prolonged (2%).
- Gastrointestinal Disorders
- Gastrointestinal disorders, predominantly diarrhea, nausea, abdominal pain and vomiting were reported frequently in the Phase III trial (see Table 1). These events began to develop primarily during the first month of treatment with SIGNIFOR and required no intervention.
- Hyperglycemia and Diabetes
- Hyperglycemia-related terms were reported frequently in the Phase III trial. For all patients, these terms included: hyperglycemia (40%), diabetes mellitus (18%), increased HbA1c (11%), and type 2 diabetes mellitus (9%). In general, increases in fasting plasma glucose (FPG) and hemoglobin A1c (HbA1c) were seen soon after initiation of SIGNIFOR and were sustained during the treatment period. In the SIGNIFOR 0.6 mg group, mean fasting plasma glucose (FPG) levels increased from 98.6 mg/dL at baseline to 125.1 mg/dL at Month 6. In the SIGNIFOR 0.9 mg group, mean fasting plasma glucose (FPG) levels increased from 97.0 mg/dL at baseline to 128.0 mg/dL at Month 6. In the SIGNIFOR 0.6 mg group, HbA1c increased from 5.8% at baseline to 7.2% at Month 6. In the SIGNIFOR 0.9 mg group, HbA1c increased from 5.8% at baseline to 7.3% at Month .
- At one month follow-up visits following discontinuation of SIGNIFOR, mean FPG and HbA1c levels decreased but remained above baseline values. Long-term follow-up data are not available.
- Elevated Liver Tests
- In the Phase III trial, there were transient mean elevations in aminotransferase values in patients treated with SIGNIFOR. Mean values returned to baseline levels by Month 4 of treatment. The elevations were not associated with clinical symptoms of hepatic disease.
- In the clinical development program of SIGNIFOR, there were 4 patients with concurrent elevations in ALT greater than 3 x ULN and bilirubin greater than 2 x ULN: one patient with Cushing’s disease and three healthy volunteers. In all four cases, the elevations were noted within the first 10 days of treatment. In all of these cases, total bilirubin elevations were seen either concomitantly or preceding the transaminase elevation. The patient with Cushing’s disease developed jaundice. All four cases had resolution of the laboratory abnormalities with discontinuation of SIGNIFOR.
- Hypocortisolism
- Cases of hypocortisolism were reported in the Phase III study in Cushing’s disease patients. The majority of cases were manageable by reducing the dose of SIGNIFOR and/or adding low-dose, short-term glucocorticoid therapy.
- Injection Site Reactions
- Injection site reactions were reported in 17% of patients enrolled in the Phase III trial in Cushing’s disease. The events were most frequently reported as local pain, erythema, hematoma, hemorrhage, and pruritus. These events resolved spontaneously and required no intervention.
- Thyroid function
- Hypothyroidism with the use of SIGNIFOR was reported for seven patients participating in the Phase III study in Cushing’s disease. All seven patients presented with a TSH close to or below the lower limit at study entry which precludes establishing a conclusive relationship between the adverse event and the use of SIGNIFOR.
- Other Abnormal Laboratory Findings
- Asymptomatic and reversible elevations in lipase and amylase were observed in patients receiving SIGNIFOR in clinical studies. Pancreatitis is a potential adverse reaction associated with the use of somatostatin analogs due to the association between cholelithiasis and acute pancreatitis.
- For hemoglobin levels, mean decreases that remained within normal range were observed. Also, post-baseline elevations in PT and PTT were noted in 33% and 47% of patients, respectively. The PT and PTT elevations were minimal.
- These laboratory findings are of unclear clinical significance.
Postmarketing Experience
There is limited information regarding Postmarketing Experience of Pasireotide in the drug label.
Drug Interactions
- Effects of Other Drugs on SIGNIFOR
- Anti-Arrhythmic Medicines and Drugs that Prolong QT
- Co-administration of drugs that prolong the QT interval with SIGNIFOR may have additive effects on the prolongation of the QT interval. Caution is required when co-administering SIGNIFOR with anti-arrhythmic medicines and other drugs that may prolong the QT interval.
- Effects of SIGNIFOR on Other Drugs
- Cyclosporine
- Concomitant administration of cyclosporine with pasireotide may decrease the relative bioavailability of cyclosporine and, therefore, dose adjustment of cyclosporine to maintain therapeutic levels may be necessary
- Bromocriptine
- Co-administration of somatostatin analogues with bromocriptine may increase the blood levels of bromocriptine. Dose reduction of bromocriptine may be necessary.
Use in Specific Populations
Pregnancy
- Pregnancy Category C
- There are no adequate and well-controlled studies in pregnant women. Reproduction studies have been performed in rats and rabbits which showed evidence of harm to the fetus due to pasireotide at therapeutic exposures. Animal reproduction studies are not always predictive of human response. This drug should be used during pregnancy only if clearly needed.
- Dosing in rats before mating and continuing into gestation at exposures less than the human clinical exposure based on body surface area comparisons across species, resulted in adverse fertility effects including: statistically significant increased implantation loss and decreased viable fetuses, corpora lutea, and implantation sites. Abnormal cycles or acyclicity were observed at systemic exposure 5-fold higher than the maximum therapeutic exposure based on surface area, comparisons across species.
- In embryofetal development studies in rats given 1, 5, and 10 mg/kg/day subcutaneously throughout organogenesis, maternal toxicity was observed at all doses, including the lowest dose tested which had exposures 4-times higher than that at the maximum therapeutic dose based on AUC comparisons across species.
- In embryofetal development studies in rabbits given 0.05, 1, and 5 mg/kg/day subcutaneously through organogenesis, maternal toxicity was observed at 1 mg/kg/day at an exposure 7-times higher than the maximum therapeutic exposure. Treatment related increased incidence of skeletal malformations were observed at 0.05 mg/kg/day, exposures less than the maximum therapeutic exposure based on AUC comparisons across species.
- In pre- and post-natal developmental studies in rats given subcutaneous doses of 2, 5, and 10 mg/kg/day during gestation through lactation and weaning, maternal toxicity was observed at all doses including the lowest dose (12-times higher than the maximum therapeutic dose based on surface area comparisons across species). Retardation of physiological growth, attributed to GH inhibition was observed at 2 mg/kg/day during a pre- and postnatal study in rats. After weaning, body weight gains in the rat pups (F1 generation) exposed to pasireotide were comparable to controls, showing reversibility of this developmental delay.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Pasireotide in women who are pregnant.
Labor and Delivery
- No data in humans are available. Studies in rats have shown no effects on labor and delivery.
Nursing Mothers
- It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when SIGNIFOR is administered to a nursing woman. Pasireotide was excreted into rat milk at levels 30% of the plasma level. As a risk to the breastfed child cannot be excluded, SIGNIFOR should not be used by the nursing mother.
Pediatric Use
- Safety and effectiveness of SIGNIFOR have not been established in pediatric patients.
Geriatic Use
- Clinical studies of SIGNIFOR did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and concomitant disease or other drug therapy.
Gender
There is no FDA guidance on the use of Pasireotide with respect to specific gender populations.
Race
There is no FDA guidance on the use of Pasireotide with respect to specific racial populations.
Renal Impairment
- No dosage adjustment of SIGNIFOR in patients with impaired renal function is required.
Hepatic Impairment
- Dose adjustment is not required in patients with mild impaired hepatic function (Child-Pugh A), but is required for patients with moderately impaired hepatic function (Child-Pugh B). Avoid the use of SIGNIFOR in patients with severe hepatic impairment (Child-Pugh C).
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Pasireotide in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Pasireotide in patients who are immunocompromised.
Administration and Monitoring
Administration
- Subcutaneous
Monitoring
There is limited information regarding Monitoring of Pasireotide in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Pasireotide in the drug label.
Overdosage
Acute Overdose
- No cases of overdosage have been reported in patients with Cushing’s disease receiving SIGNIFOR subcutaneously. Doses up to 2.1 mg b.i.d. have been used in healthy volunteers with adverse reactions of diarrhea being observed at a high frequency.
- In the event of overdosage, it is recommended that appropriate supportive treatment be initiated, as dictated by the patient’s clinical status, until resolution of the symptoms.
- Up-to-date information about the treatment of overdose can be obtained from a certified Regional Poison Center.
Chronic Overdose
There is limited information regarding Chronic Overdose of Pasireotide in the drug label.
Pharmacology

Mechanism of Action
- SIGNIFOR is an injectable cyclohexapeptide somatostatin analogue. Pasireotide exerts its pharmacological activity via binding to somatostatin receptors (ssts). Five human somatostatin receptor subtypes are known: hsst 1, 2, 3, 4, and 5. These receptor subtypes are expressed in different tissues under normal physiological conditions. Corticotroph tumor cells from Cushing’s disease patients frequently over-express hsst5 whereas the other receptor subtypes are often not expressed or are expressed at lower levels. Pasireotide binds and activates the hsst receptors resulting in inhibition of ACTH secretion, which leads to decreased cortisol secretion.
- The binding affinities of endogenous somatostatin and pasireotide are shown in Table 2.
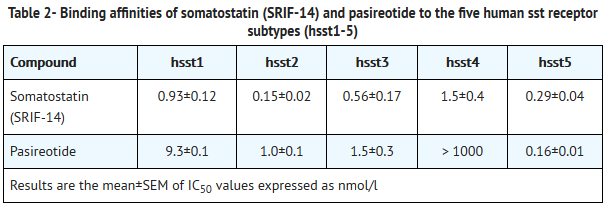
This image is provided by the National Library of Medicine.

Structure
- SIGNIFOR (pasireotide) injection is prepared as a sterile solution of pasireotide diaspartate in a tartaric acid buffer for administration by subcutaneous injection. SIGNIFOR is a somatostatin analog. Pasireotide diaspartate, chemically known as (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3-ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18-hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt, is a cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin.
- The molecular formula of pasireotide diaspartate is C58H66N10O9 • 2 C4H7NO4 and the molecular weight is 1313.41. The structural formula is:
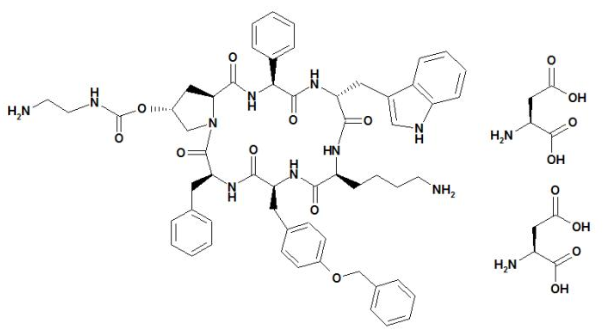
This image is provided by the National Library of Medicine.

- SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
- Each glass ampule contains:
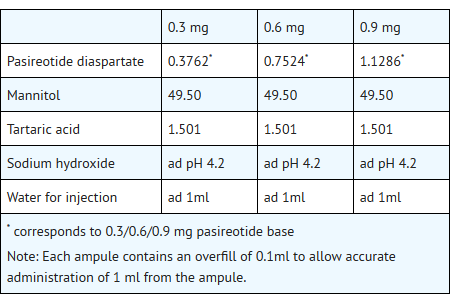
This image is provided by the National Library of Medicine.

Pharmacodynamics
- Cardiac Electrophysiology
- QTcI interval was evaluated in a randomized, blinded, crossover study in healthy subjects investigating pasireotide doses of 0.6 mg b.i.d. and 1.95 mg b.i.d. The maximum mean (95% upper confidence bound) placebo-subtracted QTcI change from baseline was 12.7 (14.7) ms and 16.6 (18.6) ms, respectively. Both pasireotide doses decreased heart rate, with a maximum mean (95% lower confidence bound) placebo-subtracted change from baseline of -10.9 (-11.9) beats per minute (bpm) observed at 1.5 hours for pasireotide 0.6 mg bid, and -15.2 (-16.5) bpm at 0.5 hours for pasireotide 1.95 mg b.i.d. The supra-therapeutic dose (1.95 mg b.i.d) produced mean steady-state Cmax values 3.3-fold the mean Cmax for the 0.6 mg b.i.d dose in the study.
Pharmacokinetics
- In healthy volunteers, pasireotide demonstrates approximately linear pharmacokinetics (PK) for a dose range from 0.0025 to 1.5 mg. In Cushing’s disease patients, pasireotide demonstrates linear dose-exposure relationship in a dose range from 0.3 to 1.2 mg.
- Absorption and Distribution:
- In healthy volunteers, pasireotide peak plasma concentration is reached within Tmax 0.25-0.5 hour. Cmax and AUC are dose-proportional following administration of single and multiple doses.
- No studies have been conducted to evaluate the absolute bioavailability of pasireotide in humans. Food effect is unlikely to occur since SIGNIFOR is administered via a parenteral route.
- In healthy volunteers, pasireotide is widely distributed with large apparent volume of distribution (Vz/F >100 L). Distribution between blood and plasma is concentration independent and shows that pasireotide is primarily located in the plasma (91%). Plasma protein binding is moderate (88%) and independent of concentration.
- Pasireotide has low passive permeability and is likely to be a substrate of P-gp (P-glycoprotein), but the impact of P-gp on ADME (absorption, distribution, metabolism, excretion) of pasireotide is expected to be low. Pasireotide is not a substrate of efflux transporter BCRP (breast cancer resistance protein), influx transporter OCT1 (organic cation transporter 1), or influx transporters OATP (organic anion-transporting polypeptide) 1B1, 1B3, or 2B1.
- Metabolism and Excretion:
- Pasireotide was shown to be metabolically stable in human liver and kidney microsomes systems. In healthy volunteers, pasireotide in its unchanged form is the predominant form found in plasma, urine and feces. Somatropin may increase CYP450 enzymes and, therefore, suppression of growth hormone secretion by somatostatin analogs including pasireotide may decrease the metabolic clearance of compounds metabolized by CYP450 enzymes.
- Pasireotide is eliminated mainly via hepatic clearance (biliary excretion) with a small contribution of the renal route. In a human ADME study 55.9 ± 6.63% of the radioactivity dose was recovered over the first 10 days post dosing, including 48.3 ± 8.16% of the radioactivity in feces and 7.63 ± 2.03% in urine.
- The clearance (CL/F) of pasireotide in healthy volunteers and Cushing’s disease patients is ~7.6 liters/h and ~3.8 liters/h, respectively.
- Steady-state pharmacokinetics:
- Following multiple subcutaneous doses, pasireotide demonstrates linear pharmacokinetics in the dose range of 0.05 to 0.6 mg once a day in healthy volunteers, and 0.3 mg to 1.2 mg twice a day in Cushing’s disease patients. Based on the accumulation ratios of AUC, the calculated effective half-life (t1/2,eff) in healthy volunteers was approximately 12 hours (on average between 10 and 13 hours for 0.05, 0.2 and 0.6 mg once a day doses).
- Special Populations:
- Population PK analyses of SIGNIFOR indicates that body weight, age, and gender do not affect pasireotide pharmacokinetics and there is no meaningful difference in pharmacokinetics between Caucasian and non-Caucasian.PK parameters.
- Hepatic impairment
- In a clinical study in subjects with impaired hepatic function (Child-Pugh A, B and C), subjects with moderate and severe hepatic impairment (Child-Pugh B and C) showed significantly higher exposures than subjects with normal hepatic function. Upon comparison with the control group, AUCinf was increased by 12%, 56% and 42% and Cmax increased by 3%, 46% and 33%, respectively, in the mild, moderate and severe hepatic impairment groups.
- Pediatric patients
- No studies have been performed in pediatric patients.
- Geriatric patients
- No clinical pharmacology studies have been performed in geriatric patients.
- Renal impairment
- Clinical pharmacology studies have not been performed in patients with impaired renal function. However, renal clearance has a minor contribution to the elimination of pasireotide in humans. Renal function is not expected to significantly impact the circulating levels of pasireotide.
- Drug Interaction Studies:
- There was no significant drug interaction between pasireotide and metformin, nateglinide or liraglutide.
Nonclinical Toxicology
- Carcinogenesis
- A life-time carcinogenicity study was conducted in rats and transgenic mice. Rats were given daily subcutaneous doses of pasireotide at 0.01, 0.05, 0.3 mg/kg/day for 104 weeks. There were no drug-related tumors in rats at exposures up to 7-fold higher than the maximum recommended clinical exposure at the 1.8 mg/day dose. Mice were given subcutaneous doses of pasireotide at 0.5, 1.0, 2.5 mg/kg/day for 26 weeks and did not identify any carcinogenic potential.
- Mutagenesis
- Pasireotide was not genotoxic in a battery of in vitro assays (Ames mutation test in Salmonella and E coli. and mutation test in human peripheral lymphocytes). Pasireotide was not genotoxic in an in vivo rat bone marrow nucleus test.
- Impairment of Fertility
- Subcutaneous dosing at 0.1 mg/kg/day before mating and continuing into gestation in rats at exposures less than the human clinical exposure based on body surface area comparisons across species resulted in statistically significant increased implantation loss and decreased viable fetuses, corpora lutea, and implantation sites. Abnormal cycles or acyclicity were observed at 1 mg/kg/day (5-fold higher than the maximum therapeutic exposure based on surface area, comparisons across species).
Clinical Studies
- A Phase III, multicenter, randomized study was conducted to evaluate the safety and efficacy of two dose levels of SIGNIFOR over a 6-month treatment period in Cushing’s disease patients with persistent or recurrent disease despite pituitary surgery or de novo patients for whom surgery was not indicated or who had refused surgery.
- Patients with a baseline 24-hour urine free cortisol (UFC) >1.5 x upper limit of normal (ULN) were randomized to receive a SIGNIFOR dosage of either 0.6 mg subcutaneous b.i.d. or 0.9 mg subcutaneous b.i.d. After three months of treatment, patients with a mean 24-hour UFC ≤ 2.0 x ULN and below or equal to their baseline values continued blinded treatment at the randomized dose until Month 6. Patients who did not meet these criteria were unblinded and the dose was increased by 0.3 mg b.i.d. After the initial six months in the study, patients entered an additional 6-month open-label treatment period. The dosage could be reduced by 0.3 mg b.i.d. at any time during the study for intolerability.
- A total of 162 patients were enrolled in this study. The majority of patients were female (78%) and had persistent or recurrent Cushing’s disease despite pituitary surgery (83%) with a mean age of 40 years. A few patients (4%) in either treatment group received previous pituitary irradiation. The median value of the baseline 24-hour UFC for all patients was 565 nmol/24 hours (normal range 30 to 145 nmol/24 hours). About two-thirds of all randomized patients completed six months of treatment.
- The primary efficacy endpoint was the proportion of patients who achieved normalization of mean 24-hour UFC levels after six months of treatment and did not dose increase during this period.
- 24-Hour Urinary Free Cortisol Results
- At Month 6, the percentages of responders for the primary endpoint were 15% and 26% in the 0.6 mg b.i.d. and 0.9 mg b.i.d. groups, respectively (Table 3). The percentages of patients with mUFC ≤ ULN or ≥ 50% reduction from baseline, a less stringent endpoint than the primary endpoint, were 34% in the 0.6 mg bid and 41% in the 0.9 mg bid groups. Dose increases appeared to have minimal effect on 24-hour UFC response. Mean and median percentage changes in UFC from baseline are presented in Table 3.
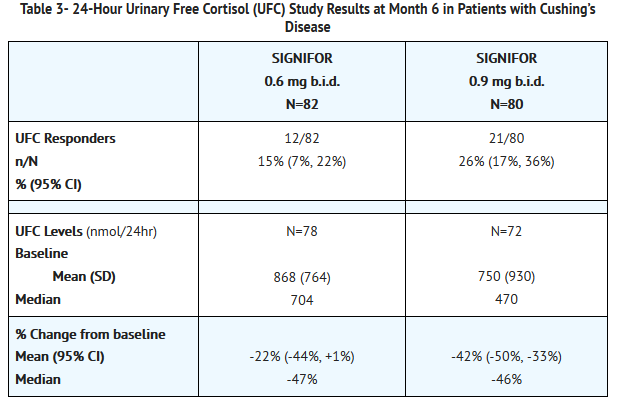
This image is provided by the National Library of Medicine.

- SIGNIFOR resulted in a decrease in the mean 24-hour UFC after 1 month of treatment (Figure 1). For patients (n=78) who stayed in the trial, similar UFC lowering was observed at Month 12.
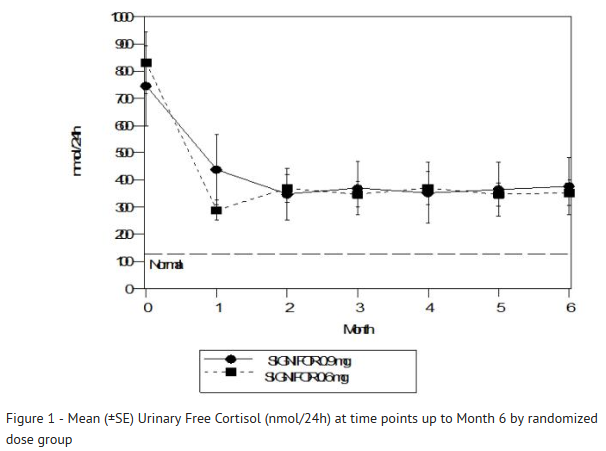
This image is provided by the National Library of Medicine.

- Note: Only patients who completed 6 months of treatment are included in this analysis (n=110). The reference line is the upper limit of normal for UFC, which is 145 nmol/24hour; +/-Standard errors are displayed.
- Other endpoints
- Decreases from baseline for blood pressure were observed at Month 6, including patients who did not receive any antihypertensive medication. However, due to the fact that the study allowed initiation of antihypertensive medication and dose increases in patients already receiving such medications, the individual contribution of SIGNIFOR or of antihypertensive medication adjustments cannot be clearly established.
- The mean decreases from baseline at Month 6 for weight, body mass index and waist circumference were 4.4 kg, 1.6 kg/m2 and 2.6 cm, respectively. Individual patients showed varying degrees of improvement in Cushing's disease manifestations but because of the variability in response and the absence of a control group in this trial, it is uncertain whether these changes could be ascribed to the effects of SIGNIFOR.
How Supplied
- SIGNIFOR is supplied as a single dose, colorless glass ampule packaged in a box of 60 ampules, arranged in 10 packs of 6 ampules each. The following packaging configurations are available.
- 0.3 mg/1 mL pasireotide (as diaspartate)
- Box of 60 ampules NDC# 0078-0633-20
- 0.6 mg/1 mL pasireotide (as diaspartate)
- Box of 60 ampules NDC# 0078-0634-20
- 0.9 mg/1 mL pasireotide (as diaspartate)
- Box of 60 ampules NDC# 0078-0635-20
- Storage and Handling
- Store at 25° C (77°F); excursions permitted to 15°-30°C (59°-86°F), protect from light.
Storage
There is limited information regarding Pasireotide Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Pasireotide |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Pasireotide |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- Counsel patients on the following possible significant adverse reactions:
- Hypocortisolism
- Hyperglycemia and diabetes
- Bradycardia and QT prolongation
- Liver test elevations
- Cholelithiasis
- Pituitary hormone deficiency
- Instruct the patients on the proper use of SIGNIFOR, including instructions to:
- Carefully review the Medication Guide.
- Do not reuse unused portions of SIGNIFOR ampules and properly dispose of the ampules after use.
- Avoid multiple injections at or near the same site within short periods of time.

This image is provided by the National Library of Medicine.

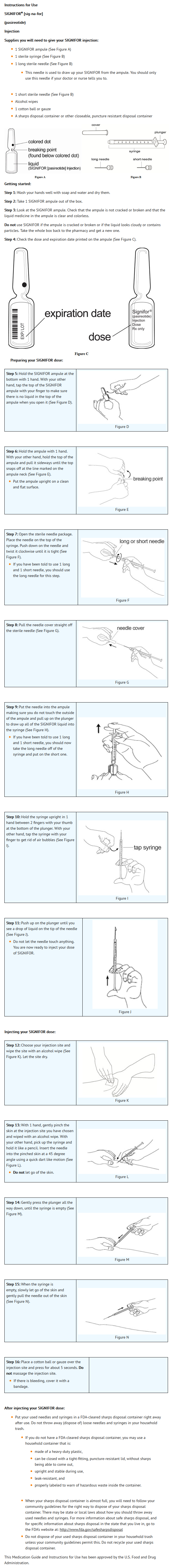
This image is provided by the National Library of Medicine.

Precautions with Alcohol
- Alcohol-Pasireotide interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- SIGNIFOR®[1]
Look-Alike Drug Names
There is limited information regarding Pasireotide Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Page Name=Pasireotide
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Pasireotide |Label Name=Pasireotide06.png
}}
{{#subobject:
|Label Page=Pasireotide |Label Name=Pasireotide07.png
}}
{{#subobject:
|Label Page=Pasireotide |Label Name=Pasireotide08.png
}}
{{#subobject:
|Label Page=Pasireotide |Label Name=Pasireotide09.png
}}