Cystic fibrosis pathophysiology: Difference between revisions
No edit summary |
|||
| Line 85: | Line 85: | ||
</div> | </div> | ||
==Microscopic Pathology== | ==Microscopic Pathology== | ||
<div align="left"> | <div align="left"> | ||
<gallery heights="175" widths="175"> | <gallery heights="175" widths="175"> | ||
Revision as of 15:34, 8 March 2018
| https://https://www.youtube.com/watch?v=BhFpFiZumS0%7C350}} |
|
Cystic fibrosis Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Cystic fibrosis pathophysiology On the Web |
|
American Roentgen Ray Society Images of Cystic fibrosis pathophysiology |
|
Risk calculators and risk factors for Cystic fibrosis pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Shaghayegh Habibi, M.D.[2]
Overview
Cystic fibrosis is an autosomal recessive disease that caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Substitution of a single amino acid is the most common type of CFTR gene mutation. CFTR gene functions as a chloride channel (pumps chloride from the intracellular space to the extracellular space) found on the surface of the epithelial cells. The genetic mutations result in defective transport of chloride, and secondarily sodium and eventually abnormal viscous mucoid secretions mostly in lungs (results in airway surface liquid depletion, decreased mucociliary transport, inflammation and infection) and GI tract (results in reduced volume of pancreatic secretion, pancreatic tissue destruction and fibrosis, malnutrition and poor growth). Infertility due to atresia/absent vasa deferentia and abnormal/absent seminal vesicles is the associated condition of cystic fibrosis.
Pathophysiology
Pathogenesis
Cystic fibrosis (CF) is an autosomal recessive disease that caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR gene functions as a chloride channel (pumps chloride from the intracellular space to the extracellular space) found on the surface of the epithelial cells that line multiple organs especially lungs and GI tract. The genetic mutations result in defective transport of chloride, and secondarily sodium, by epithelial cells and eventually abnormal viscous mucoid secretions mostly in lungs and GI tract. Other organs containing epithelia such as the sweat glands, biliary duct, the male reproductive tract and the intestine are also affected. Two mechanisms which cause airway-surface-liquid depletion are as follow:[1][2][3]
| Lack of CFTR normal activity | |||||||||||||||||||||
| Less chloride secretion | More sodium absorption | ||||||||||||||||||||
| Less water transport into the epithelial surface layer | Excessive sodium and water absorption through the epithelial channel | ||||||||||||||||||||
lung involvement in cystic fibrosis
In patients with cystic fibrosis abnormal chloride conductance of epithelial cells results in airway surface liquid depletion and decreased mucociliary transport (airway surface liquid is essential to support ciliary functioning). The consequence of this is a vicious circle of inflammation, tissue damage and infection.[3]
Also exaggerated, generalized and prolonged inflammatory response of lungs to bacterial and viral pathogen is observed. The inflammatory response is characterised by neutrophil dominated airway inflammation which is present even in clinically stable patients and in young infants diagnosed by neonatal screening.[3][4] Breakdown of accumulated neutrophils in the infected lungs of patients with cystic fibrosis leads to the release of large amounts of DNA. Accumulated DNA causes high viscosity of the infected sputum, followed by decreased ciliary transport and function.[5]
Gastrointestinal tract involvement in cystic fibrosis
Pancreatic disease:
In cystic fibrosis about 90% of patients present with exocrine pancreatic insufficiency. Pancreatic disease results from a reduced volume of pancreatic secretion with low concentrations of HCO3, followed by retained and prematurely activated digestive proenzymes in pancreatic ducts, result in tissue destruction and fibrosis.[6] Abnormally viscous secretions in the ducts of the pancreas, followed by loss of pancreatic exocrine function results in malnutrition and poor growth.[4]
Biliary disorders:
In one third of patients with cystic fibrosis have abnormal results of liver function tests. Fatty infiltration is reported in up to 70% of older patients and in nearly 10% of these it progresses to biliary cirrhosis. Histological evaluations show duct dilatation and intraluminal concretions. Bile-duct epithelium becomes hyperplastic with periductal inflammation and fibrosis. In up to one third of patients with cystic fibrosis a small and poorly functioning gallbladder is detected.[6]
Genetics

Cystic fibrosis is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. This gene codes for a chloride transporter regulated by cyclic AMP (cAMP)-dependent phosphorylation. There are almost 2,000 variants of CFTR gene mutations:[4]
- Substitution of a single amino acid (40%)
- Alter RNA processing including nonsense, frameshift and missplicing (36%)
- Large rearrangements of CFTR (3%)
- Promoter regions (1%)
- Neutral variants (14%)
- The effect of the remaining 6% is unclear.
In Caucasian populations, the frequency of mutations is as follows:[8]Template:Entête tableau charte alignement
! Mutation
! Frequency
worldwide
|-----
| ΔF508
| 66.0%
|-
| G542X
| 2.4%
|-----
| G551D
| 1.6%
|-
| N1303K
| 1.3%
|-----
| W1282X
| 1.2%
|}
Children who inherit one mutated CFTR gene and one normal CFTR gene are "CF carriers". CF carriers usually have no symptoms of cystic fibrosis but they can pass the mutated CFTR gene to their children.[9]
Associated Conditions
- In cystic fibrosis 98% of men are infertile. The causes of aspermia include:[10]
- Atresia or absent vasa deferentia
- abnormal or absent seminal vesicles
Gross Pathology
-
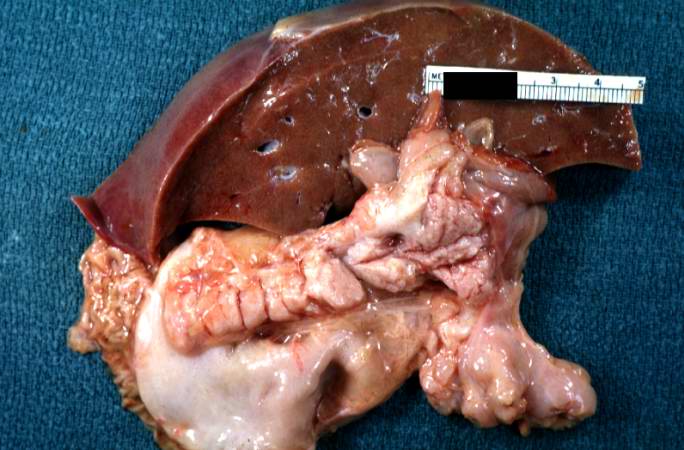
A gross photograph of liver and pancreas from the autopsy. The pancreas is slightly smaller than normal and it has a mucous consistency.
-
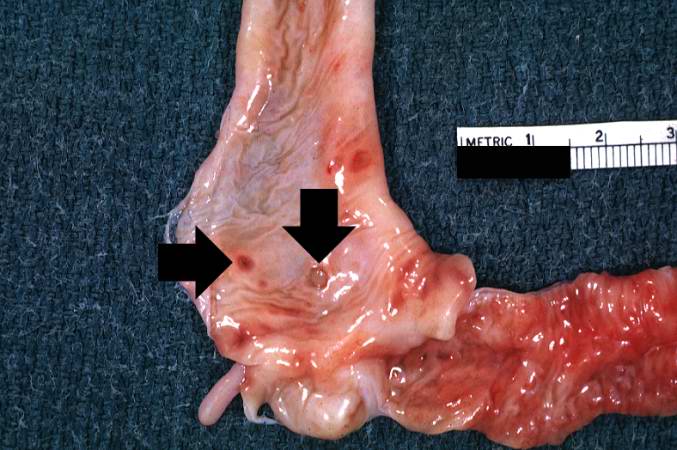
This section of duodenum demonstrates dilation, loss of rugae, and areas of ulceration (arrows).


Microscopic Pathology
-
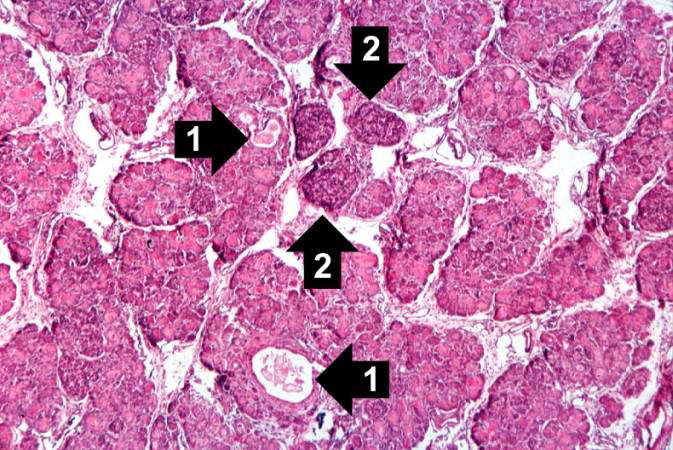
This higher-power photomicrograph of the pancreas shows interstitial tissue and the presence of small cystic spaces (1) within the acinar lobules. These spaces are filled with an eosinophilic proteinaceous material. The islets of Langerhans (2) are unaffected.
-
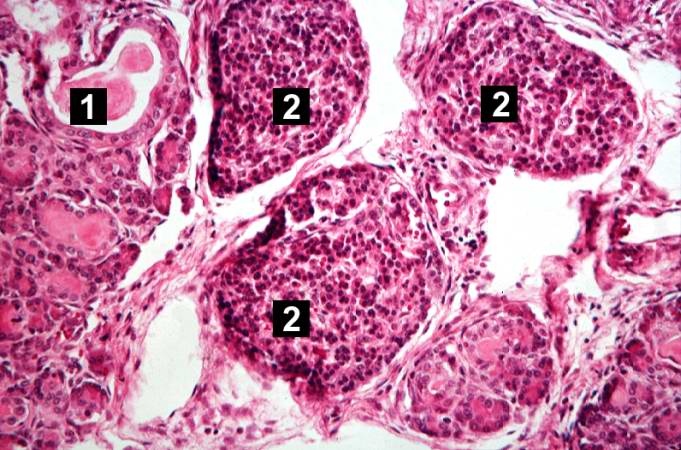
This higher-power photomicrograph shows a cystic space (1) within an acinar lobule. Islets of Langerhans (2) are also visible.
-
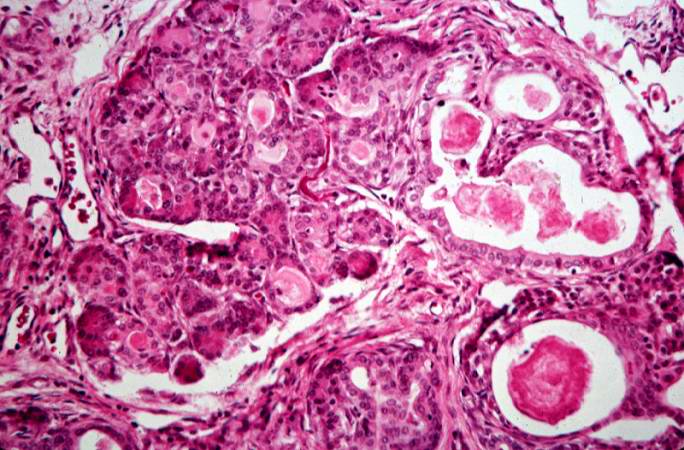
This high-power photomicrograph shows more clearly these variably-sized cystic spaces within the acinar pancreas.



-
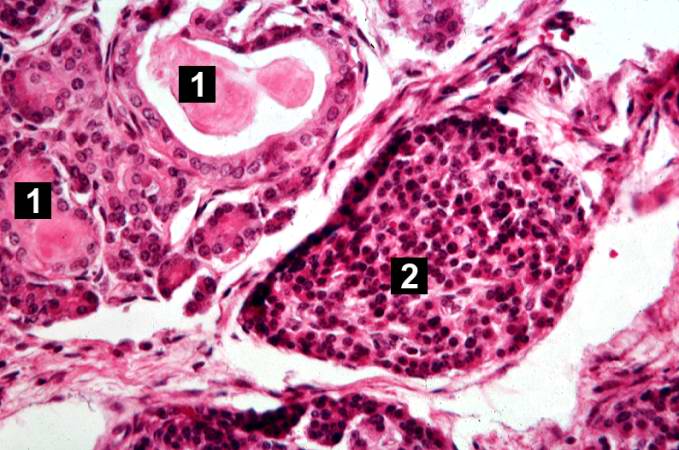
This is another high-power photomicrograph showing cystic spaces (1) within the acinar pancreas and a normal islet of Langerhans (2).
-
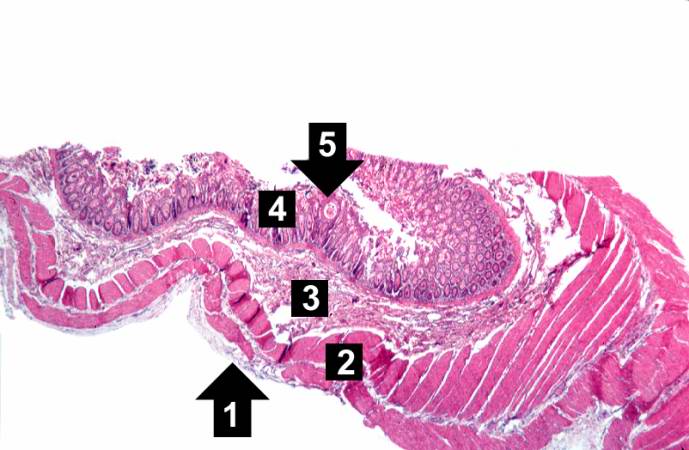
This low-power photomicrograph of intestine shows the normal layers of the intestine, including the serosa (1), the muscularis (2), the submucosa (3), and the mucosal layer (4) with its deep mucosal crypts. There is yet another cystic space within the mucosa (5).
-
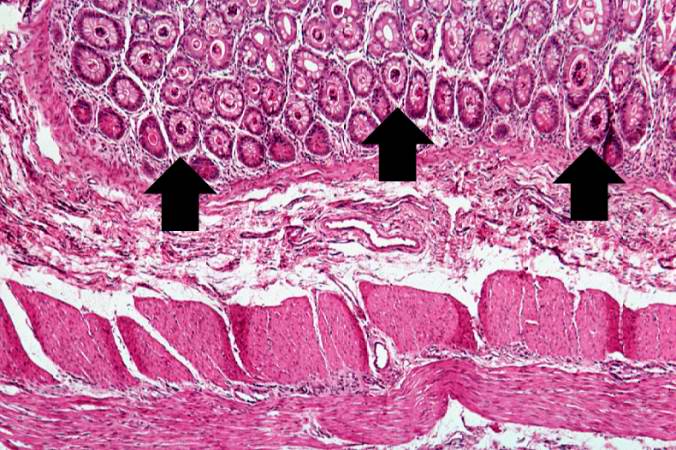
A higher-power photomicrograph shows the bottom of the intestinal crypts and the other normal layers of the intestine. Even at this magnification, accumulations of eosinophilic debris can be seen in many of the intestinal crypts (arrows).



-
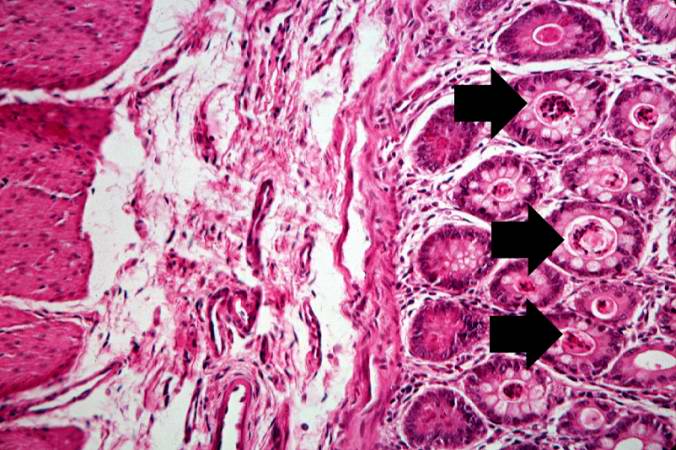
This is a higher-power photomicrograph showing the eosinophilic debris in many of the intestinal crypts (arrows).
-
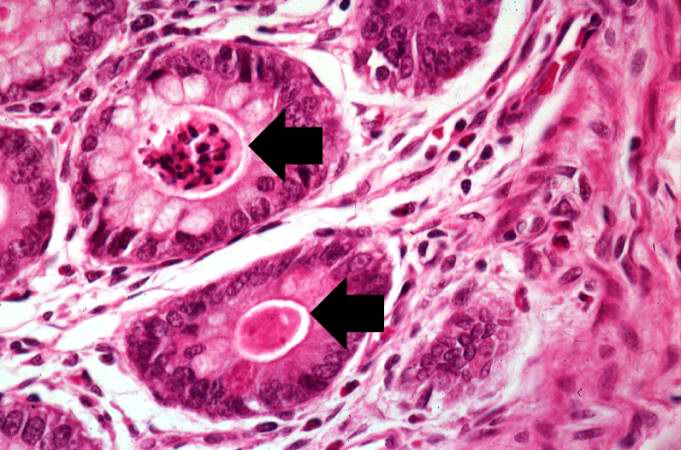
This higher-power photomicrograph shows more clearly the eosinophilic debris (arrows) in the intestinal crypts.
-
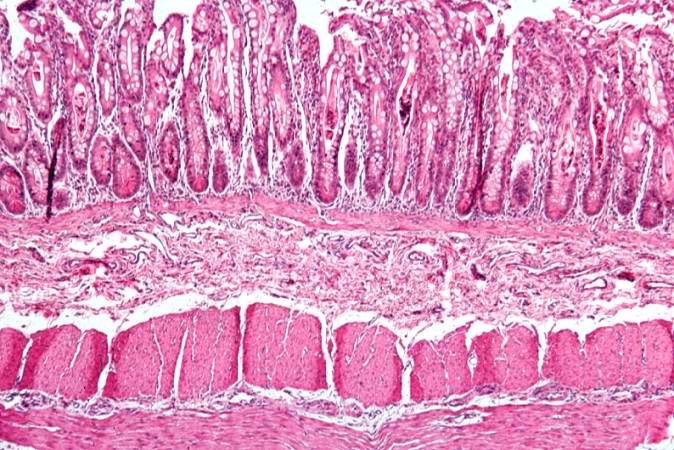
This is a low-power photomicrograph from another section of the intestine. Saggital sections of the intestinal crypts show the crypts along their full length, extending to the mucosal surface.



-
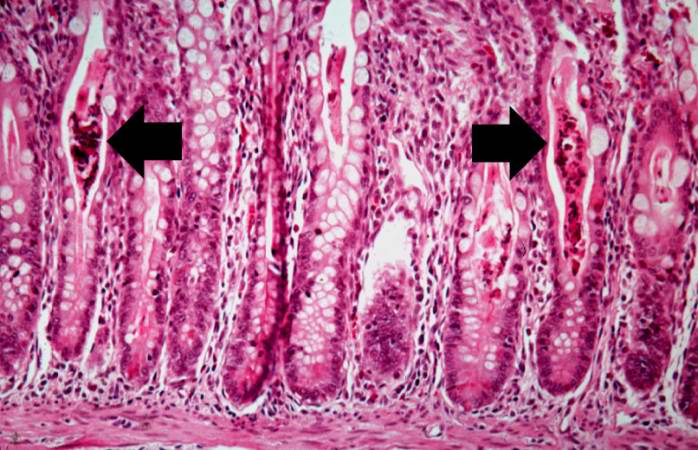
A higher-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows).
-
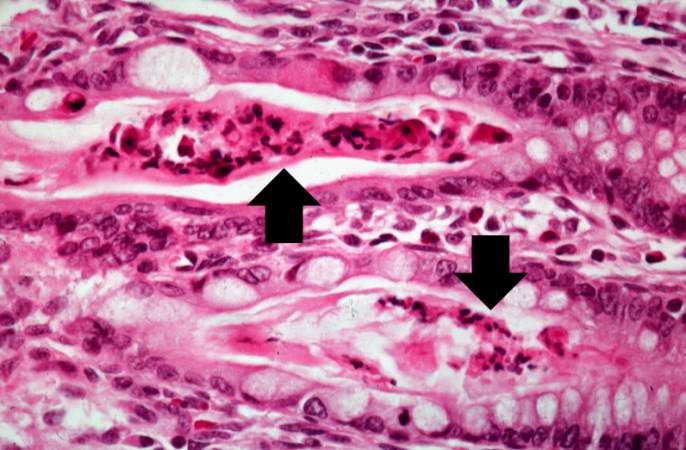
Another high-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows).
-
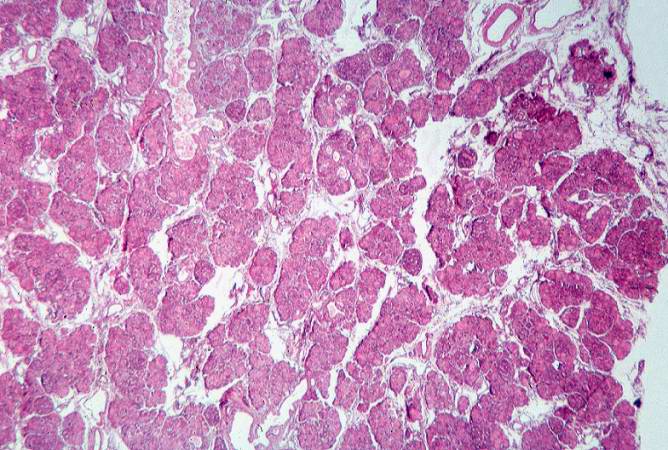
This low-power photomicrograph of pancreas shows increased interstitial connective tissue resulting in accentuation of the lobular pattern.



References
- ↑ National Center for Biotechnology Information (US). Genes and Disease [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1998-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22183/
- ↑ Voter KZ, Ren CL (2008). "Diagnosis of cystic fibrosis". Clin Rev Allergy Immunol. 35 (3): 100–6. doi:10.1007/s12016-008-8078-x. PMID 18506640.
- ↑ 3.0 3.1 3.2 Ratjen FA (2009). "Cystic fibrosis: pathogenesis and future treatment strategies". Respir Care. 54 (5): 595–605. PMID 19393104.
- ↑ 4.0 4.1 4.2 Cutting GR (2015). "Cystic fibrosis genetics: from molecular understanding to clinical application". Nat. Rev. Genet. 16 (1): 45–56. doi:10.1038/nrg3849. PMC 4364438. PMID 25404111.
- ↑ Konstan MW, Ratjen F (2012). "Effect of dornase alfa on inflammation and lung function: potential role in the early treatment of cystic fibrosis". J. Cyst. Fibros. 11 (2): 78–83. doi:10.1016/j.jcf.2011.10.003. PMC 4090757. PMID 22093951.
- ↑ 6.0 6.1 Ratjen F, Döring G (2003). "Cystic fibrosis". Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
- ↑ "File:CFTR.jpg - Wikimedia Commons". External link in
|title=(help) - ↑ Prevalence of ΔF508, G551D, G542X, R553X mutations among cystic fibrosis patients in the North of Brazil. Brazilian Journal of Medical and Biological Research 2005; 38:11–15. PMID 15665983
- ↑ "Cystic Fibrosis - National Library of Medicine - PubMed Health".
- ↑ Ratjen F, Döring G (2003). "Cystic fibrosis". Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185.
{kind=link}