Systemic lupus erythematosus pathophysiology: Difference between revisions
No edit summary |
|||
| Line 40: | Line 40: | ||
|HLA genes | |HLA genes | ||
| colspan="1" rowspan="1" |DR2, DR3, DR4, DR7, DR8, DRw12 DQw2, DQA1, DQB1, DQ6, DQw6, DQ7, DQw7, DQw8, DQw9 B61 , B8 | | colspan="1" rowspan="1" |DR2, DR3, DR4, DR7, DR8, DRw12 DQw2, DQA1, DQB1, DQ6, DQw6, DQ7, DQw7, DQw8, DQw9 B61 , B8 | ||
| | |Anticardiolipin or lupus anticoagulant) | ||
Anti-Sm | |||
Anti-U1 ribonuclear protein | |||
Anti-DNA | |||
Anti-La | |||
Anti-Ro | |||
|- | |- | ||
|Complement genes | |||
| colspan="1" rowspan="1" |C2, C4, C1q | |||
| | | | ||
|- | |- | ||
|Non-HLA genes | |Non-HLA genes | ||
Revision as of 20:37, 16 June 2017
|
Systemic lupus erythematosus Microchapters |
|
Differentiating Systemic lupus erythematosus from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Systemic lupus erythematosus pathophysiology On the Web |
|
American Roentgen Ray Society Images of Systemic lupus erythematosus pathophysiology |
|
Directions to Hospitals Treating Systemic lupus erythematosus |
|
Risk calculators and risk factors for Systemic lupus erythematosus pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Cafer Zorkun, M.D., Ph.D. [2] Raviteja Guddeti, M.B.B.S. [3]
Overview
Pathophysiology
- Template Sentence 1: [Pathogen name] is usually transmitted via the [transmission route] route to the human host.
- Template Sentence 2: Following transmission/ingestion, the [pathogen] uses the [entry site] to invade the [cell name] cell.
- Template Sentence 3: On gross pathology, [feature1], [feature2], and [feature3] are characteristic findings of [disease name].
- Template Sentence 4: On microscopic histopathological analysis, [feature1], [feature2], and [feature3] are characteristic findings of [disease name].
- Template Sentence 5: [Disease name] is transmitted in [mode of genetic transmission] pattern.
- Template Sentence 6: [Disease/malignancy name] arises from [cell name]s, which are [cell type] cells that are normally involved in [function of cells].
- Template Sentence 7: Development of [disease name] is the result from multiple genetic mutations.
- Template Sentence 8: Genes involved in the pathogenesis of [disease name] include [gene1], [gene2], and [gene3].
- Template Sentence 9: The progression to [disease name] usually involves the [molecular pathway].
- Template Sentence 10: The pathophysiology of [disease name] depends on the histological subtype.
Pathogenesis
Genetics
- Polygenic inheritance of the disease:
- Increase of disease occurrence in identical twins, the increase in frequency of SLE among first degree relatives, and the increased risk of developing the disease in siblings of SLE patients.
- Homozygous deficiencies of the early components of complement are associated with developing SLE or a lupus-like disease. C1q and systemic lupus erythematosus. Walport MJ, Davies KA, Botto M Immunobiology. 1998 Aug; 199(2):265-85.
- It is estimated that at least four mutated genes are needed for the development of the disease???????????????????
- major histocompatibility complex (MHC)
- the susceptibility to SLE involves human leucocyte antigen (HLA) class II gene polymorphisms
- HLA DR2 and DR3 with SLE is a common: associated with the presence of certain autoantibodies such as anti-Sm (small nuclear ribonuclear protein), anti-Ro, anti-La, anti-nRNP (nuclear ribonuclear protein), and anti-DNA antibodies/ HLA class III genes, particularly those encoding complement components C2 and C4, confer risk for SLE in certain ethnic groups
- genes that encode mannose binding protein (MBP), tumour necrosis factor α, the T cell receptor, interleukin 6 (IL-6), CR1, immunoglobulin Gm and Km allotypes, FcγRIIA and FcγRIIIA (both IgG Fc receptors), and heat shock protein 70. enetics of systemic lupus erythematosus. Clinical implications. Sullivan KE Rheum Dis Clin North Am. 2000 May; 26(2):229-56, v-vi.
- he FcγRIIA polymorphism has been associated with nephritis in African Americans and Koreans. FcgammaRIIa/IIIa polymorphism and its association with clinical manifestations in Korean lupus patients. Yun HR, Koh HK, Kim SS, Chung WT, Kim DW, Hong KP, Song GG, Chang HK, Choe JY, Bae SC, Salmon JE, Yoo DH, Kim TY, Kim SY Lupus. 2001; 10(7):466-72.
- FcγRIIIA polymorphism with SLE in Hispanics and white populations. Low-binding alleles of Fcgamma receptor types IIA and IIIA are inherited independently and are associated with systemic lupus erythematosus in Hispanic patients. Zuñiga R, Ng S, Peterson MG, Reveille JD, Baethge BA, Alarcón GS, Salmon JE Arthritis Rheum. 2001 Feb; 44(2):361-7.
- mutations of codon 54 of the MBL gene carry a minor risk for SLE susceptibility in southern Chinese. Association of systemic lupus erythematosus with promoter polymorphisms of the mannose-binding lectin gene. Ip WK, Chan SY, Lau CS, Lau YL Arthritis Rheum. 1998 Sep; 41(9):1663-8.
- Polygenic inheritance of the disease:
| HLA genes | DR2, DR3, DR4, DR7, DR8, DRw12 DQw2, DQA1, DQB1, DQ6, DQw6, DQ7, DQw7, DQw8, DQw9 B61 , B8 | Anticardiolipin or lupus anticoagulant)
Anti-Sm Anti-U1 ribonuclear protein Anti-DNA Anti-La Anti-Ro |
| Complement genes | C2, C4, C1q | |
| Non-HLA genes | Mannose binding lectin polymorphisms | |
| Tumour necrosis factor α | ||
| T cell receptor | ||
| Interleukin 6 | ||
| CR1 | ||
| Immunoglobulin Gm and Km | ||
| FcγRIIA (IgG Fc receptor) | ||
| FcγRIIIA (IgG Fc receptor) | ||
| PARP (poly-ADP ribose polymerase) | ||
| Heat shock protein 70 | ||
| Humhr 3005 | ||
Associated Conditions
- ==Gross Pathology==
- ==Microscopic Pathology==
Lupus is a chronic autoimmune disease in which the body's own defense system attacks otherwise healthy tissue. The body's immune system produces antibodies against itself, particularly against proteins in the cell nucleus. Because of genetic variations in different components of the immune system, in some people the immune system attacks these nuclear-related proteins and produces antibodies against them. In the end, these antibody complexes damage blood vessels in critical areas of the body, such as the glomeruli of the kidney; these antibody attacks are the cause of SLE.
SLE is a chronic inflammatory disease believed to be a type III hypersensitivity response with potential type II involvement.[1]
Clinically, it can affect multiple organ systems including the heart, skin, joints, kidneys and nervous system. The exact mechanisms for the development of systemic lupus erythematosus (SLE) are still unclear since the pathogenesis is a multifactorial event.
Genetics
The first mechanism may arise genetically. Research indicates that SLE may have a genetic link. Lupus does run in families, but no single "lupus gene" has yet been identified. Instead, multiple genes appear to influence a person's chance of lupus developing when triggered by environmental factors. Researchers are now identifying the individual genes, the proteins they produce, and their role in the immune system. Each protein is a link on the autoimmune chain, and researchers are trying to find drugs to break each of those links. [2][3][4]
- The most important genes are located on chromosome 6, where mutations may occur randomly (de novo) or be inherited.
- Additionally, people with SLE have an altered RUNX-1 binding site, which may be either cause or contributor (or both) to the condition. Altered binding sites for RUNX-1 have also been found in people with psoriasis and rheumatoid arthritis.
Other abnormalities include:
- Increased expression of FcεRIγ, which replaces the sometimes deficient TCR ζ chain
- Increased and sustained calcium levels in T cells
- Moderate increase of inositol triphosphate
- Reduction in PKC phosphorylation
- Increased desire of animal protein intake.
- Reduction in Ras-MAP kinase signaling
- Deficiencies in protein kinase A I activity
Environmental triggers
The second mechanism may be due to environmental factors. These factors may not only exacerbate existing lupus conditions, but also trigger the initial onset. They include:
- Certain medications (such as some antidepressants and antibiotics)
- Extreme stress
- Exposure to sunlight
- Hormones
- Infections - some researchers have sought to find a connection between certain infectious agents (viruses and bacteria), but no pathogen can be consistently linked to the disease.
- UV radiation has been shown to trigger the photosensitive lupus rash, but some evidence also suggests that UV light is capable of altering the structure of the DNA, leading to the creation of autoantibodies.
- Some researchers have found that women with silicone gel-filled breast implants have produced antibodies to their own collagen, but it is not known how often these antibodies occur in the general population and there is no data that show these antibodies cause connective tissue diseases such as lupus.
Abnormalities in apoptosis
- Apoptosis is increased in monocytes and keratinocytes
- Expression of Fas by B cells and T cells is increased
- There are correlations between the apoptotic rates of lymphocytes and disease activity
Tangible body macrophages (TBMs) are large phagocytic cells in the germinal centers of secondary lymph nodes. They express CD68 protein. These cells normally engulf B cells which have undergone apoptosis after somatic hypermutation. In some patients with SLE, significantly fewer TBMs can be found, and these cells rarely contain material from apoptotic B cells. Also, uningested apoptotic nuclei can be found outside of TBMs. This material may present a threat to the tolerization of B cells and T cells. Dendritic cells in the germinal center may endocytose such antigenic material and present it to T cells, activating them. Also, apoptotic chromatin and nuclei may attach to the surfaces of follicular dendritic cells and make this material available for activating other B cells which may have randomly acquired self-specificity through somatic hypermutation.[5]
Clearance deficiency

Beside discussed causations, impaired clearance of dying cells is a potential pathway for the development of this systemic autoimmune disease. This includes deficient phagocytic activity, scant serum components in addition to increased apoptosis.
Monocytes isolated from whole blood of SLE patients show reduced expression of CD44 surface molecules involved in the uptake of apoptotic cells. Most of the monocytes and tingible body macrophages (TBM), which are found in the germinal centres of lymph nodes, even show a definitely different morphology in patients with SLE. They are smaller or scarce and die earlier. Serum components like complement factors, CRP and some glycoproteins are furthermore decisively important for an efficiently operating phagocytosis. In patients these components are often missing, diminished or inefficient.
The clearance of early apoptotic cells is an important function in multicellular organisms. It leads to a progression of the apoptosis process and finally to secondary necrosis of the cells, if this ability is disturbed. Necrotic cells release nuclear fragments as potential autoantigens as well as internal danger signals, inducing maturation of dendritic cells (DC), since they have lost their membranes integrity. Increased appearance of apoptotic cells also is simulating inefficient clearance. That leads to maturation of DC and also to the presentation of intracellular antigens of late apoptotic or secondary necrotic cells, via MHC molecules.
Autoimmunity possibly results by the extended exposure to nuclear and intracellular autoantigens derived from late apoptotic and secondary necrotic cells. B and T cell tolerance for apoptotic cells is abrogated and the lymphocytes get activated by these autoantigens; inflammation and the production of autoantibodies by plasma cells is initiated. A clearance deficiency in the skin for apoptotic cells has also been observed in patients with cutaneous lupus erythematosus (CLE).
Accumulation in germinal centres (GC)

In healthy conditions apoptotic lymphocytes are removed in germinal centres by specialised phagocytes, the tingible body macrophages (TBM); that’s why no free apoptotic and potential autoantigenic material can bee seen.
In some patients with SLE accumulation of apoptotic debris can be observed in GC, because of an ineffective clearance of apoptotic cells.
In close proximity to TBM, follicular dendritic cells (FDC) are localized in GC, which attach antigen material to their surface and in contrast to bone marrow-derived DC, neither take it up nor present it via MHC molecules.
Autoreactive B cells can accidentally emerge during somatic hypermutation and migrate into the GC light zone. Autoreactive B cells, maturated coincidently, normally don’t receive survival signals by antigen planted on follicular dendritic cells and perish by apoptosis.
In the case of clearance deficiency apoptotic nuclear debris accumulates in the light zone of GC and gets attached to FDC. This serves as a germinal centre survival signal for autoreactive B-cells.
After migration into the mantle zone autoreactive B cells require further survival signals from autoreactive helper T cells, which promote the maturation of autoantibody producing plasma cells and B memory cells.
In the presence of autoreactive T cells a chronic autoimmune disease may be the consequence.
Drug reactions
Drug-induced lupus erythematosus is a reversible condition that usually occurs in patients being treated for a long-term illness. Drug-induced lupus mimics systemic lupus. However, symptoms of drug-induced lupus generally disappear once a patient is taken off the medication which triggered the episode.
Anti-nRNP autoimmunity
Autoantibodies to nRNP A and nRNP C initially targeted restricted, proline-rich motifs. Antibody binding subsequently spread to other epitopes. The similarity and cross-reactivity between the initial targets of nRNP and Sm autoantibodies identifies a likely commonality in cause and a focal point for intermolecular epitope spreading.[6]
Others
Elevated expression of HMGB1 was found in the sera of patients and mice with systemic lupus erythematosus, High Mobility Group Box 1 (HMGB1) is a nuclear protein participating in chromatin architecture and transcriptional regulation. Recently, there is increasing evidence that HMGB1 contributes to the pathogenesis of chronic inflammatory and autoimmune diseases due to its pro-inflammatory and immunostimulatory properties.[7]
Gross Images
-
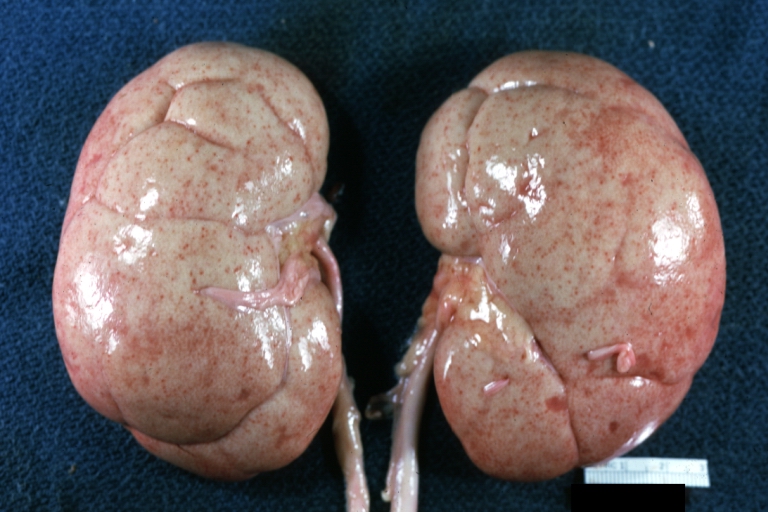
Kidney: Lupus erythematosus: Gross, enlarged very pale kidneys with flea bite or ectasia. A good example of kidneys from a patient with nephrotic syndrome (subacute glomerulonephritis)
-
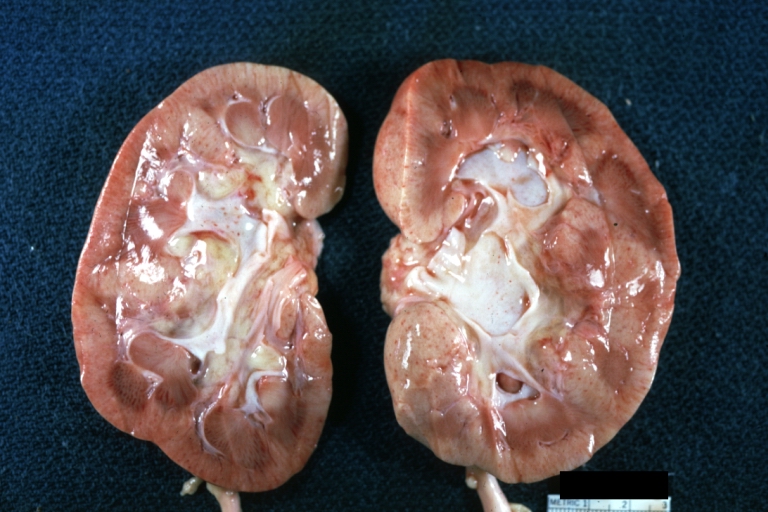
Kidney: Lupus erythematosus: Gross cut surface pale kidneys typical of nephrotic syndrome (subacute glomerulonephritis)
-
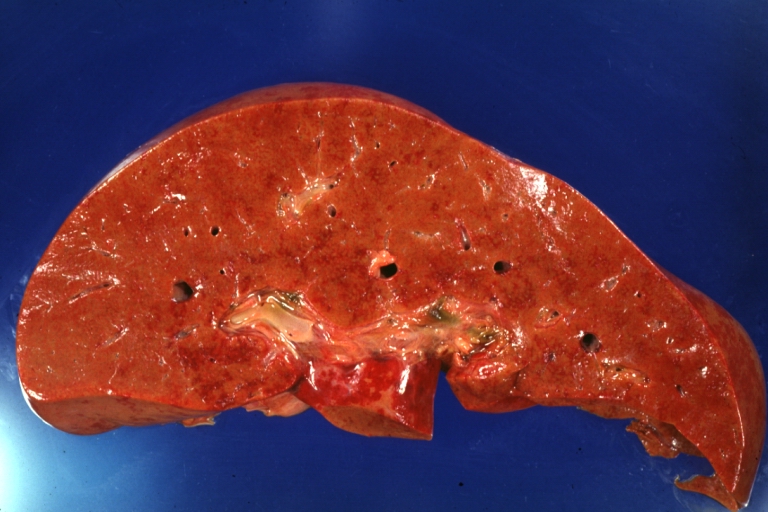
Lupus Erythematosus Hepatitis: Gross natural color. A 19yo female with lupus erythematosus and hepatitis characterized by periportal cell necrosis and sinus thrombosis cause uncertain photo shows focal grid-like hyperemia
-
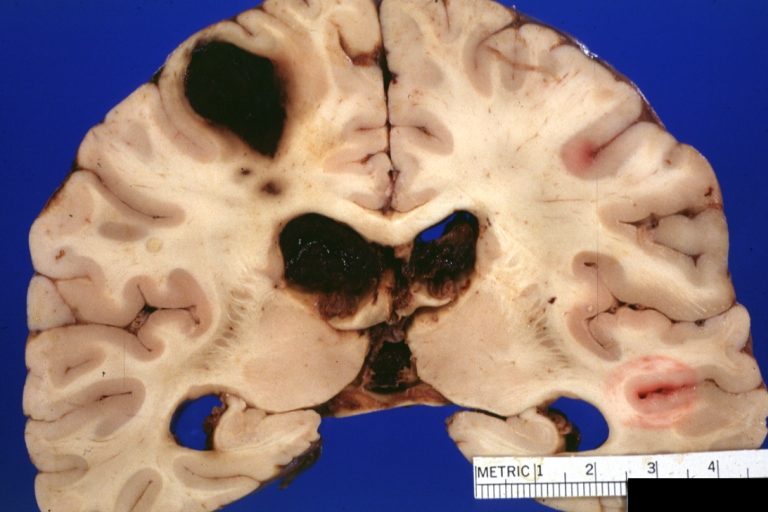
Brain: Lupus Erythematosus Libman Sacks Embolism: Gross fixed tissue one large and two small hemorrhages 19yo female with history of TIAs
-
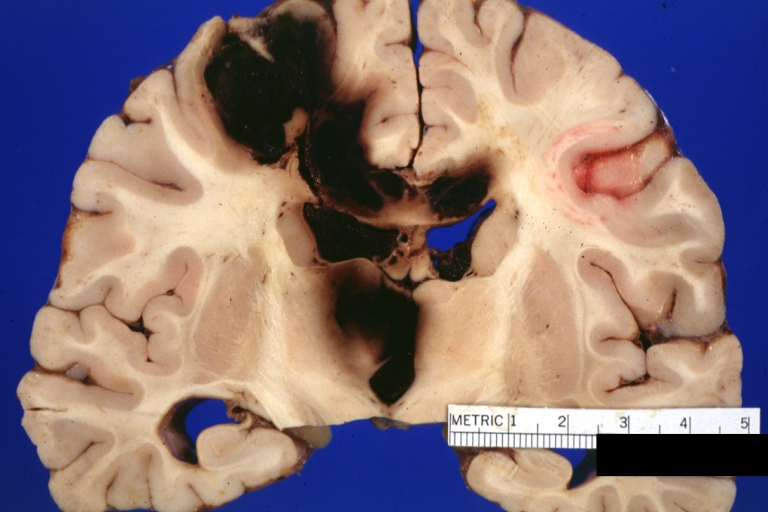
Brain: Lupus Erythematosus Libman Sacks Embolism: Gross fixed tissue large hemorrhagic infarcts due to embolism 19yo female with known lupus and history of TIAs
-
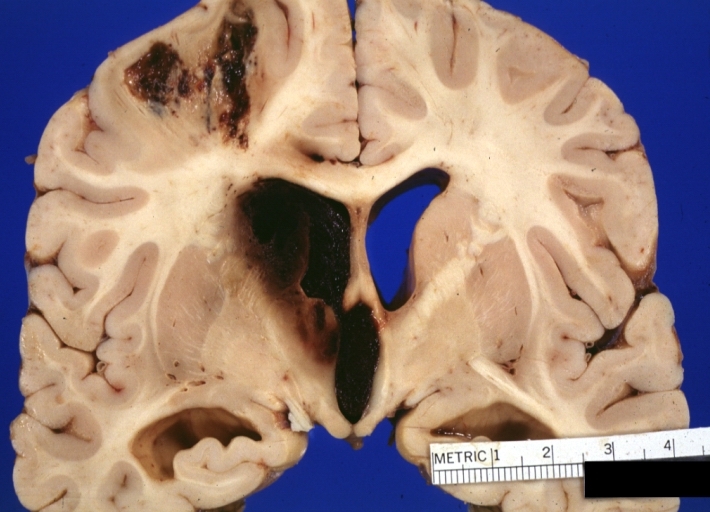
Brain: Lupus Erythematosus Libman Sacks Embolism: Gross large hemorrhagic infarcts 19 yo female with history of TIAs proved case lupus with renal failure
-
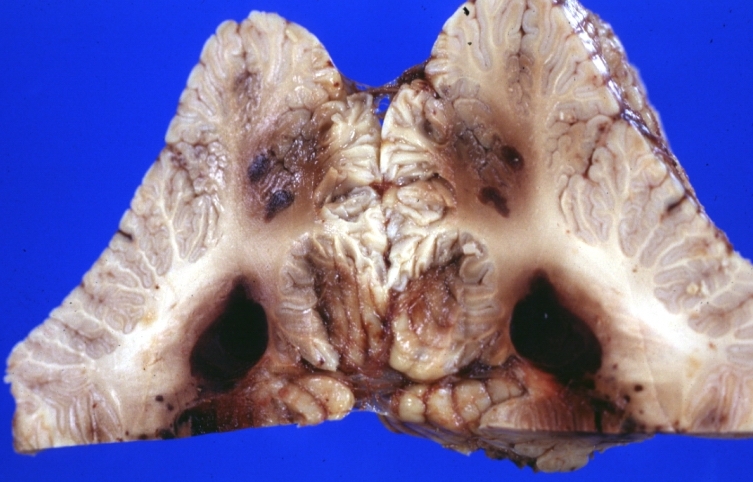
Brain: Lupus Erythematosus Libman Sacks Embolism: Gross fixed tissue cerebellar hemorrhagic infarcts 19yo female with history of TIAs known lupus case with renal failure
-
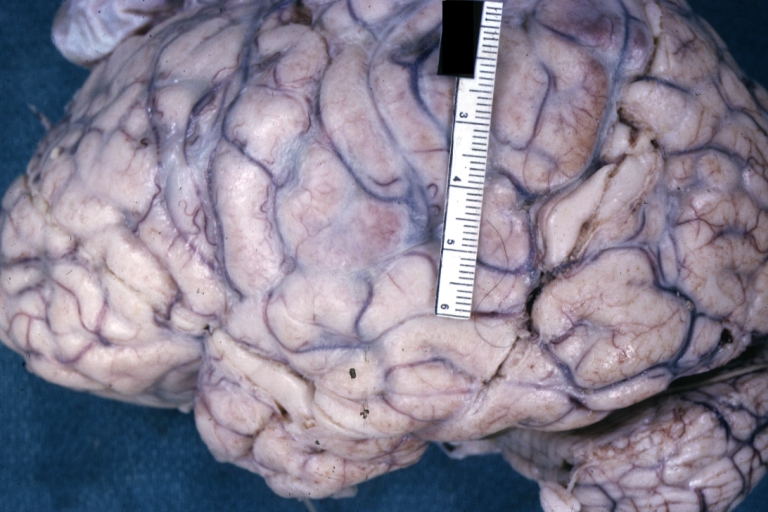
Brain: Purulent Meningitis: Gross fixed tissue excellent example Pneumococcus case of young girl with lupus
-
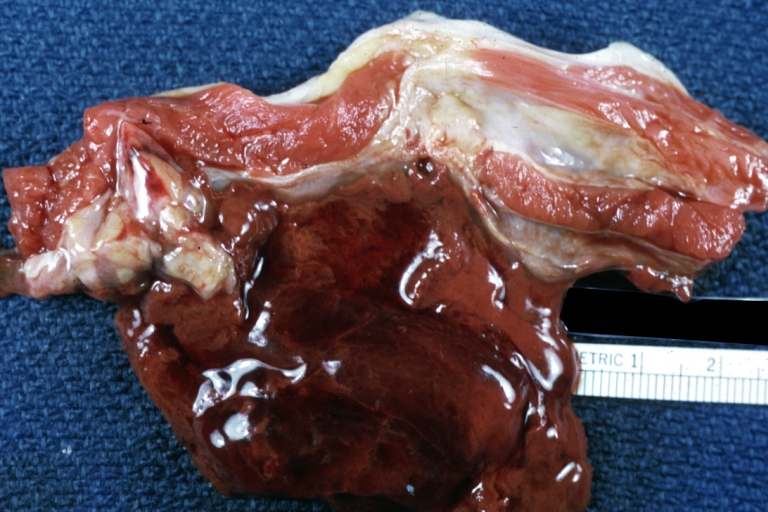
Skeletal muscle: Hematoma: Gross natural color flank muscle hematoma old showing typical chocolate appearance of blood coagulum young female with lupus and thrombocytopenia
-
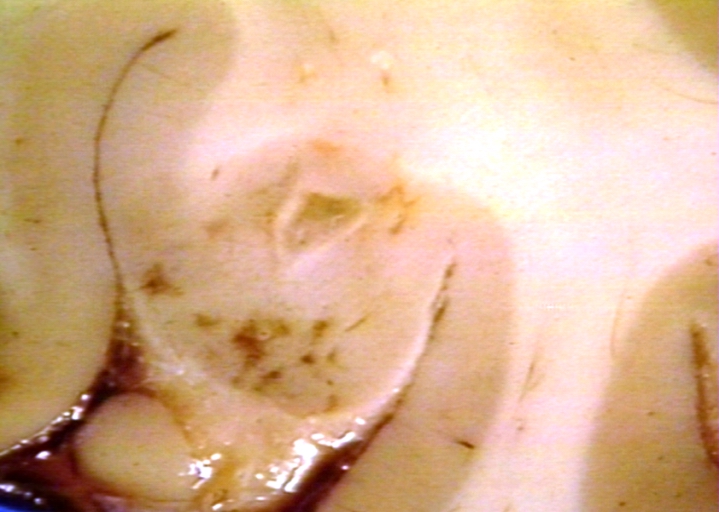
Brain: Lupus Erythematosus, Systemic; Microinfarct in Cerebral Cortex
-
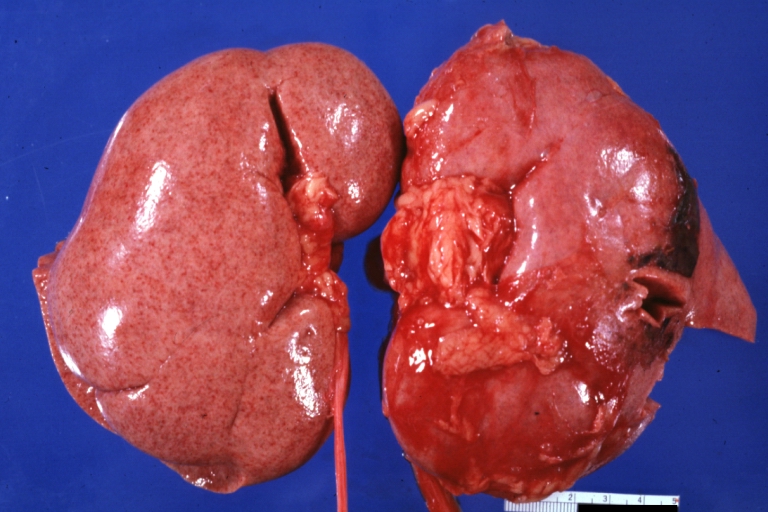
Kidney: Lupus Nephritis: Gross natural color external view of flea bitten kidneys quite good advanced proliferative type glomerulonephritis of lupus in a 16yo female
-
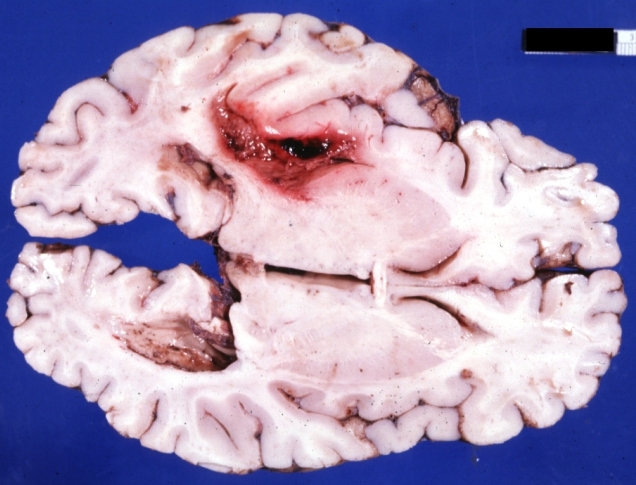
Brain: Hemorrhage Massive With Lupus Erythematosus: Gross apparently fresh tissue large left frontoparietal hemorrhagic infarct in a 16yo female with advanced lupus nephritis and sepsis
-
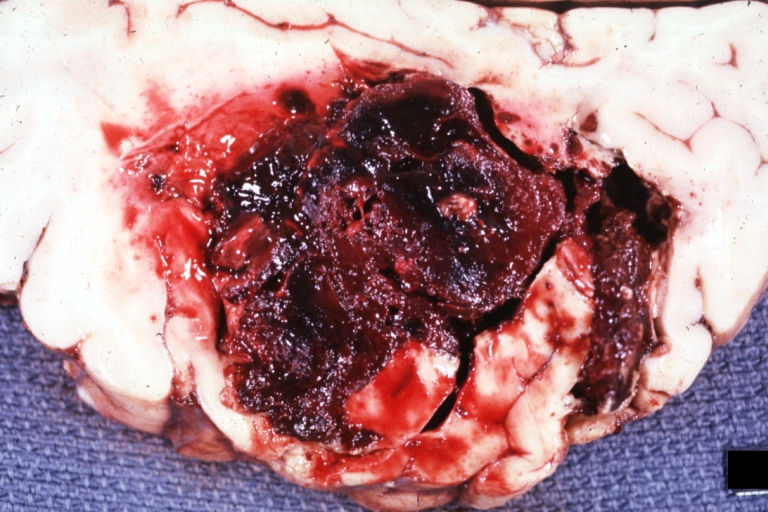
Brain: Hemorrhage Massive With Lupus Erythematosus: Gross natural color not the best exposure but OK large left frontoparietal hemorrhage in 16yo female with advanced lupus nephritis and sepsis
-
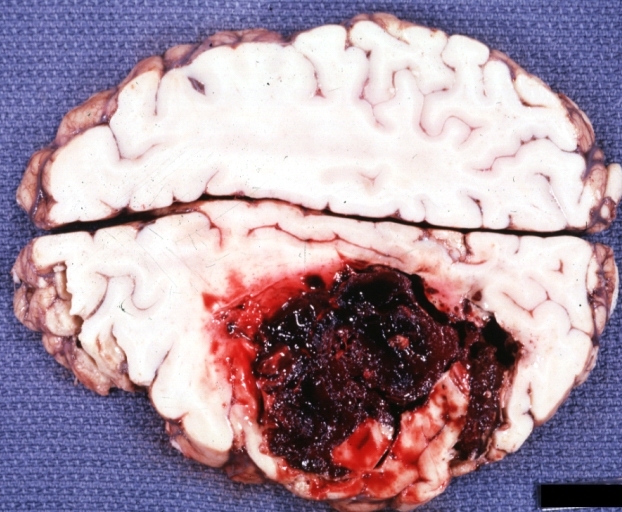
Brain: Hemorrhage Massive With Lupus Erythematosus: Gross natural color but not best exposure large left frontoparietal hemorrhage in 16yo female with advanced lupus nephritis and sepsis
-
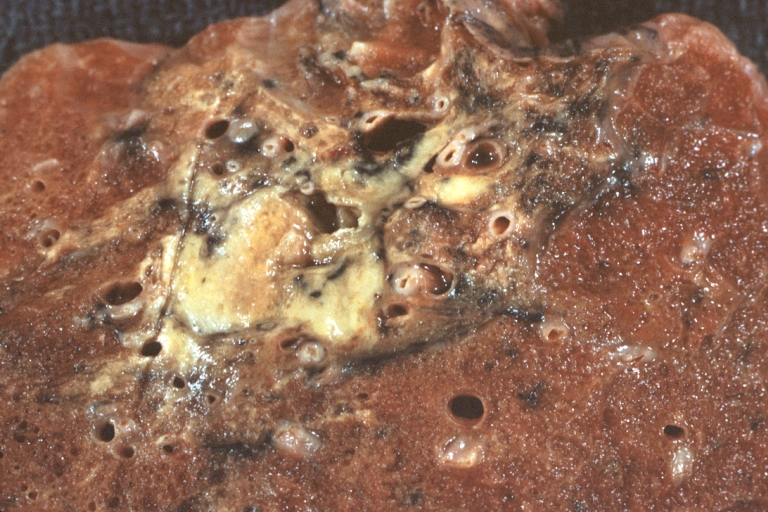
Lung: Tuberculosis Reactivation: Gross fixed tissue close-up of lung hilum with node and parenchyma lesions case of lupus erythematosus treated with prednisone for many years
-

Lung: Tuberculosis Reactivation: Gross fixed tissue close-up of lung showing lesions in nodes and parenchyma case of lupus erythematosus treated for long period with prednisone
-
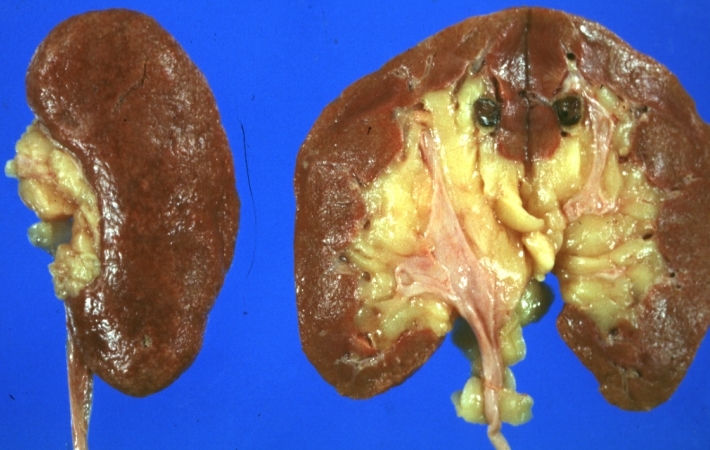
Kidney: Lupus Erythematosus: Gross natural color nice external and cut surface view of uniformly scarred and moderately shrunken kidneys
-
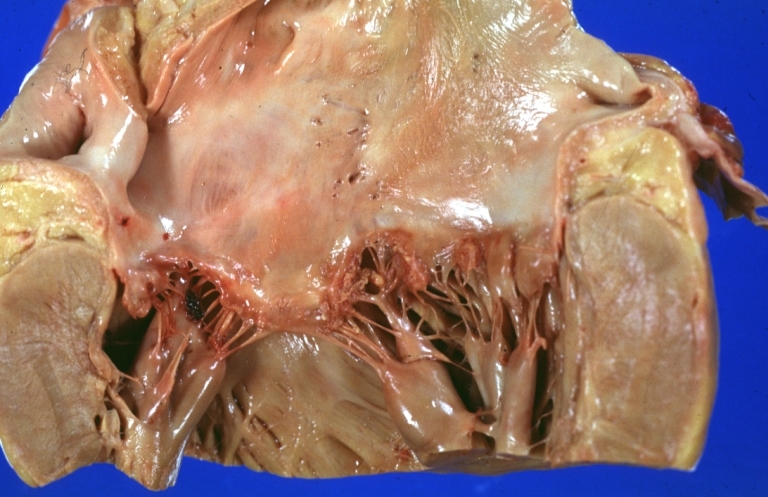
Lupus Erythematosus Libman Sacks Endocarditis: Gross natural color mitral valve small lesions but cause much trouble in form of TIAs and terminally multiple hemorrhagic brain infarcts
-
Kidney: Lupus Erythematosus: Micro high mag H&E increased mesangial tissue and wire loops 10yo female with renal failure and TIAs due to Libman Sacks endocarditis



















Microscopic Images
-
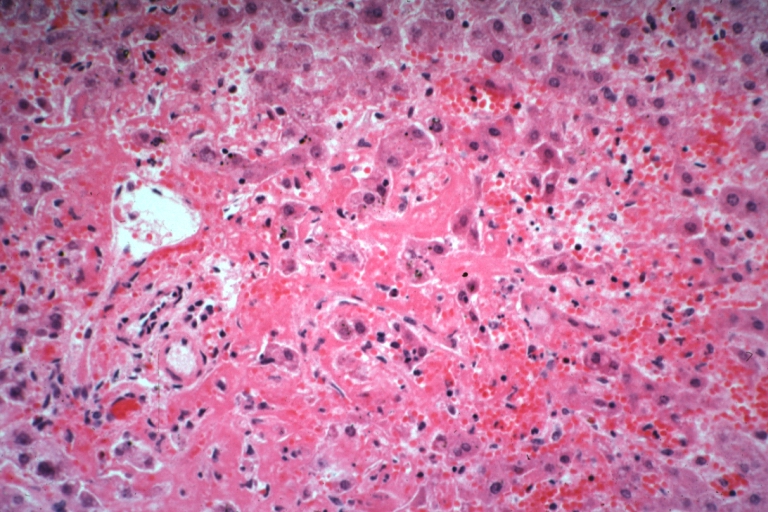
Lupus erythematosus hepatitis: Micro high mag H&E, periportal sinus thrombosis with liver cell necrosis and noninflammatory infiltrate (possibly viral). A 19yo female with lupus erythematosus
-
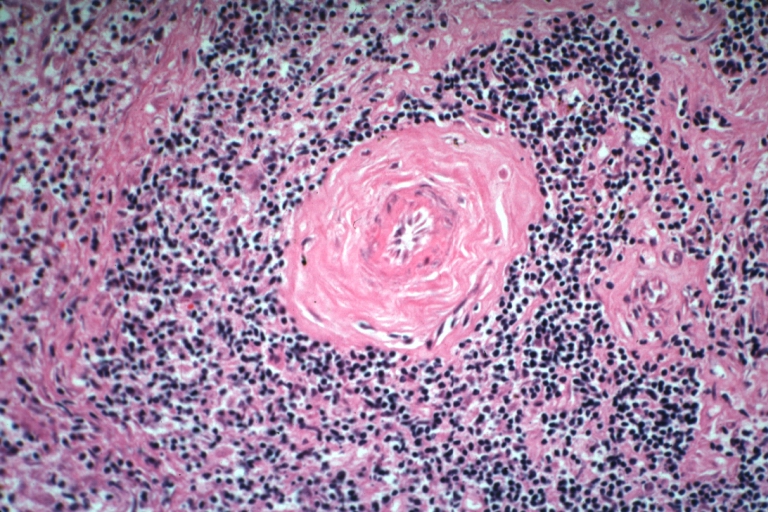
Spleen: Lupus erythematosus Periarterial Fibrosis: Micro high may H&E. An excellent example of periarterial fibrosis
-
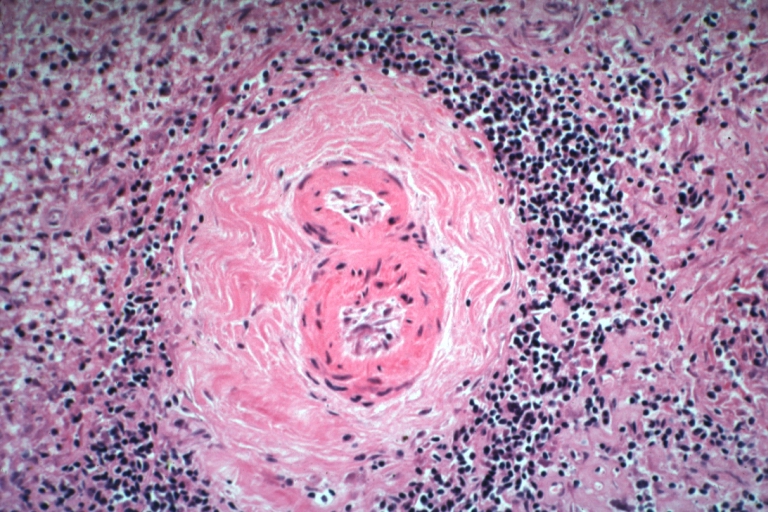
Spleen: Lupus erythematosus, periarterial fibrosis: Micro high may H&E. An excellent example of periarterial fibrosis
-
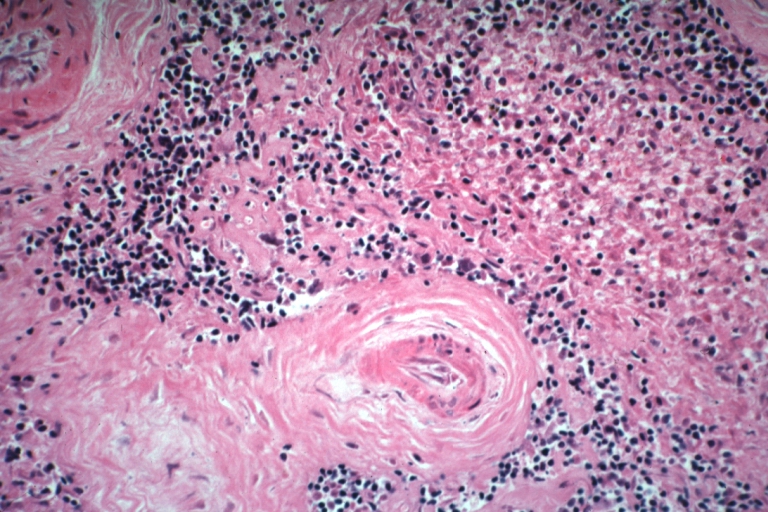
Spleen: Lupus erythematosus. Basophilic bodies and periarterial fibrosis: Micro high mag, H&E. Two basophilic bodies and periarterial fibrosis. An excellent example of this rarely seen lupus lesion.
-
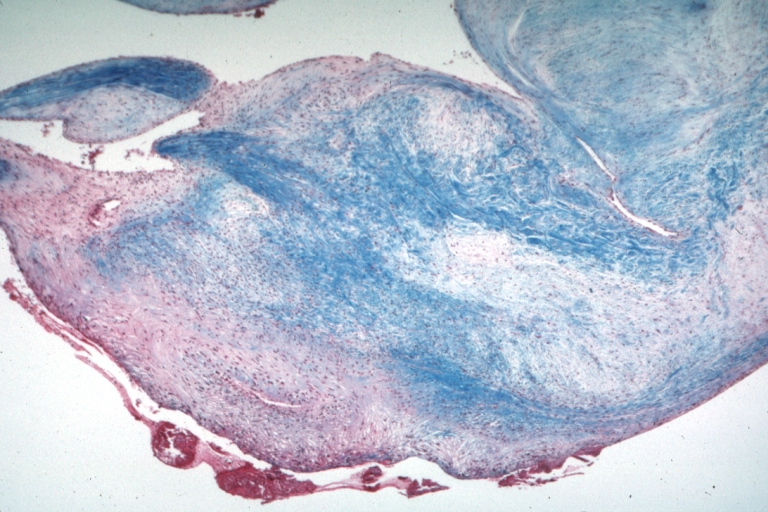
Lupus Erythematosus, Libman Sacks Endocarditis: Micro low mag trichrome stain thickened valve leaflet with small mural fibrin deposit. A 19yo female with cerebral lupus in form of TIAs due to this lesion.
-
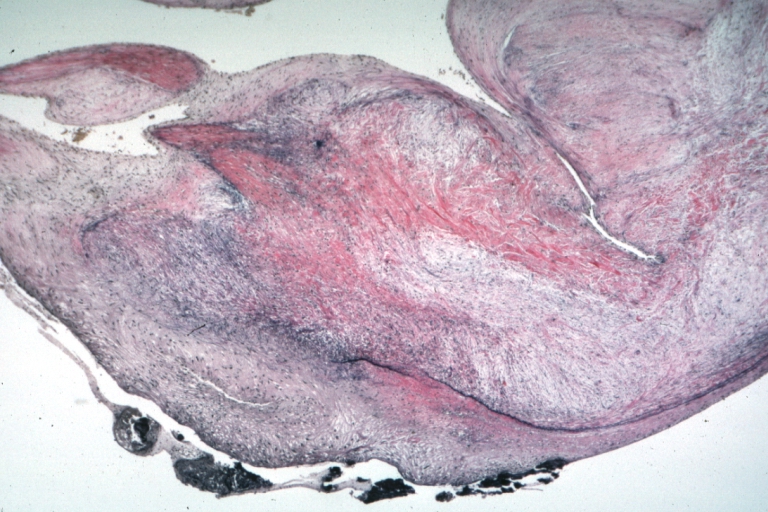
Lupus Erythematosus, Libman Sacks Endocarditis: Micro low mag, elastic van Gieson stain, mitral valve thickened, leaflet with small mural fibrin deposit that caused TIAs in 19yo female
-
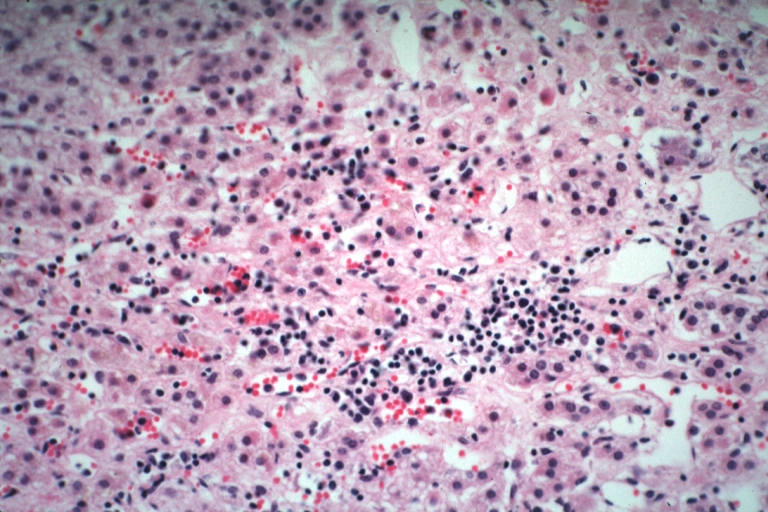
Adrenal: Autoimmune Adrenalitis: Micro high mag H&E focal area of lymphocytic infiltration in zona reticularis in a 19yo female with lupus erythematosus
-
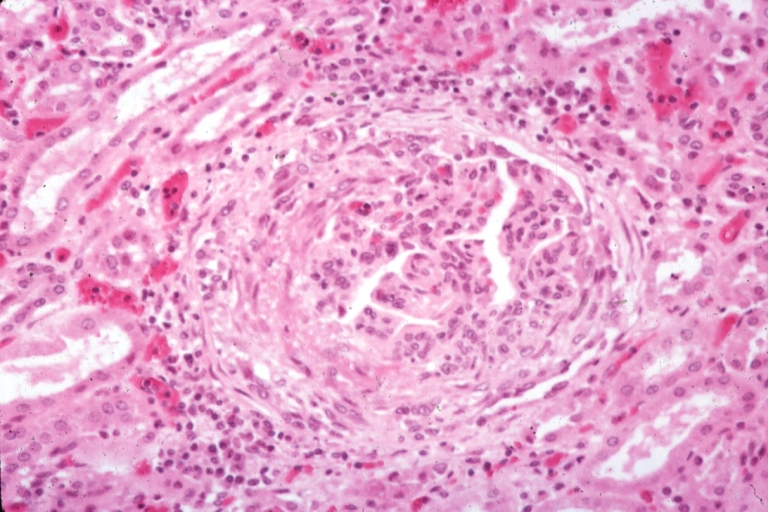
Kidney: Lupus Erythematosus: Micro high mag H&E. A nice example of a lesion of chronic glomerulonephritis with lobular scarring. A fibrous type crescent.
-
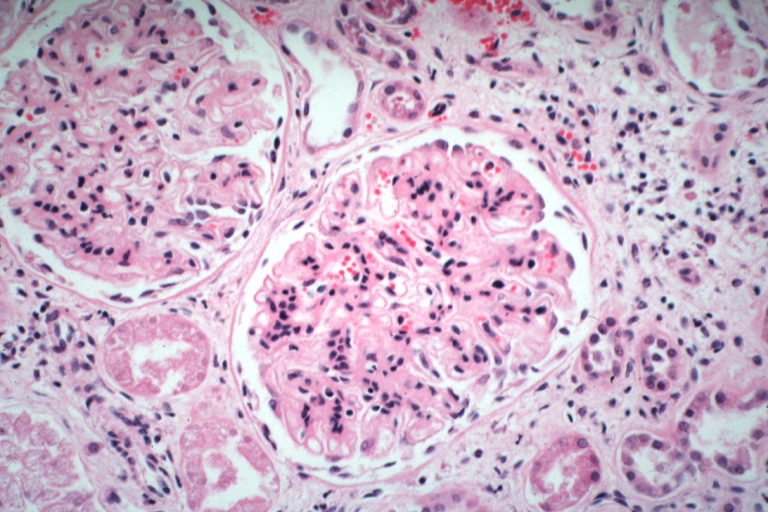
Kidney: Lupus Erythematosus: Micro high mag H&E two glomeruli showing mesangial thickening and focal wire loop type lesions 19yo female with renal failure and embolic brain disease from Libman Sacks lesion on mitral valve
-
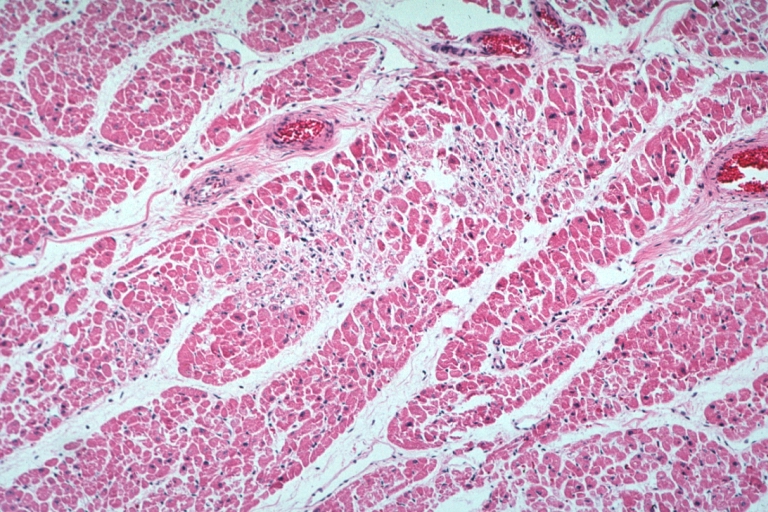
Lupus Erythematosus Myocardial Necrosis Due To Libman Sacks: Micro low mag H&E focal myocardial necrosis due to embolism from Libman Sacks lesion on mitral valve 19yo female with TIAs due to mitral lesion
-
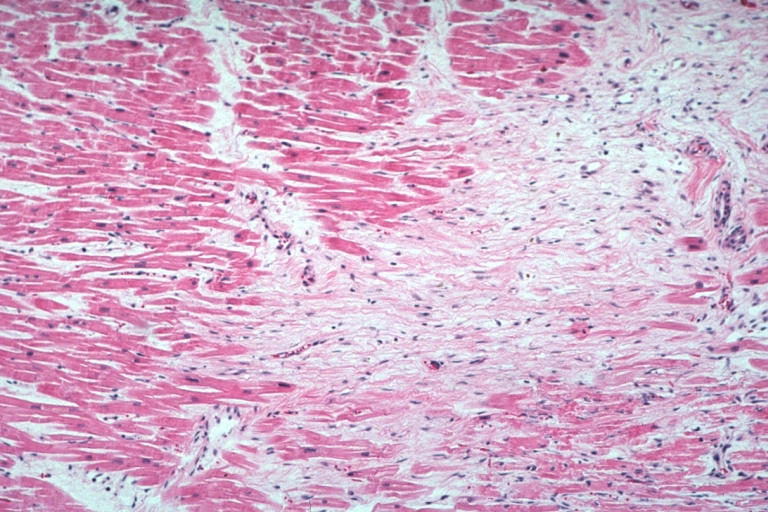
Lupus Erythematosus Focal Myocardial Scar Due To Libman Sacks Embolism: Micro low mag H&E focal scar in myocardium due to embolism
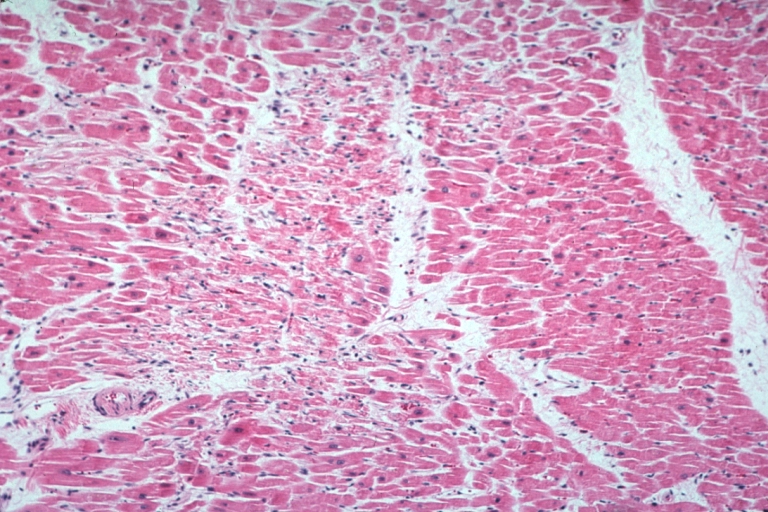
-
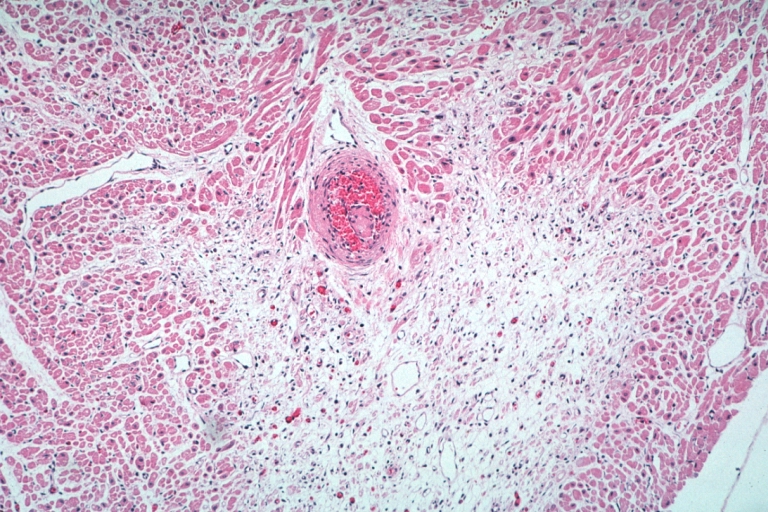
Lupus Erythematosus Myocardial Scar Due To Libman Sacks Embolism: Micro low mag H&E scar with portion of embolus in small artery
-
Lupus Erythematosus Myocardial Necrosis Due To Libman Sacks: Micro low mag H&E well shown focal myocardial necrosis due to embolism from mitral Libman Sacks lesion
-
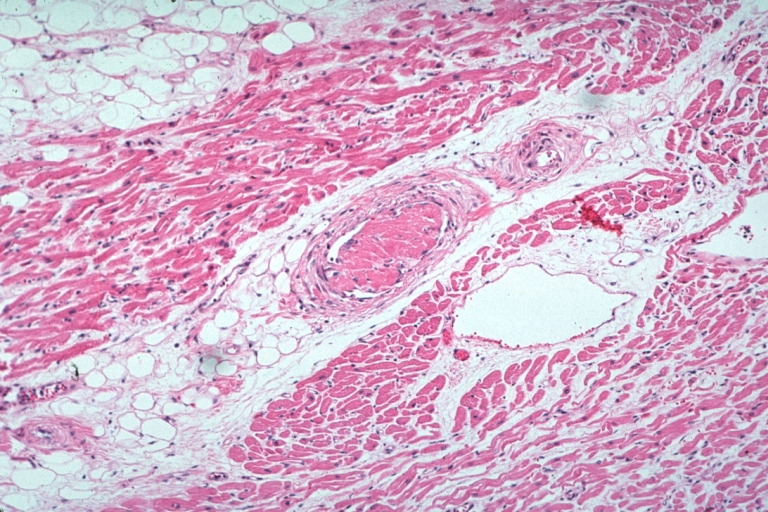
Lupus Erythematosus Embolus From Libman Sacks Lesion: Micro med mag H&E well shown embolus in small artery
-
Lupus Erythematosus Hepatitis: Micro low mag trichrome stain periportal liver cell necrosis and sinus thrombosis with no inflammatory reaction cause unknown 19yo female with lupus erythematosus
-
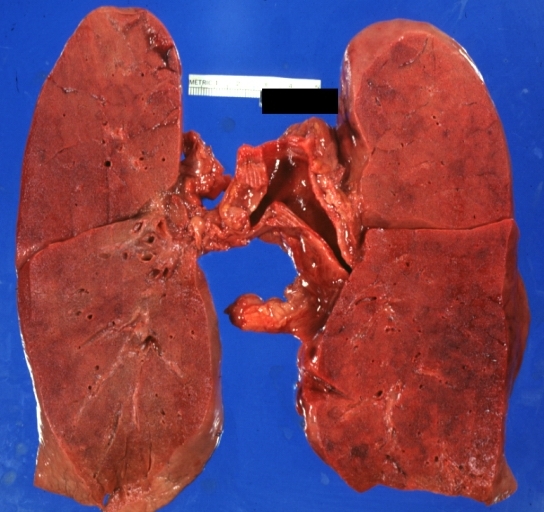
Lung: Diffuse Alveolar Damage: Gross natural color section of both lungs with frank meaty appearance case of lupus erythematosus in 19yo female
-
Lung: Necrotizing Bronchiolitis: Micro low mag H&E well shown lesion in lung that grossly looked like diffuse alveolar damage which indeed has lesions of this type additionally 19yo female with lupus erythematosus
-
Lupus Erythematosus Libman Sacks Endocarditis: Micro low mag H&E mitral valve lesion with easily seen mural thrombi and focal necrobiosis of collagen in thickened valve leaflet 19yo female
-
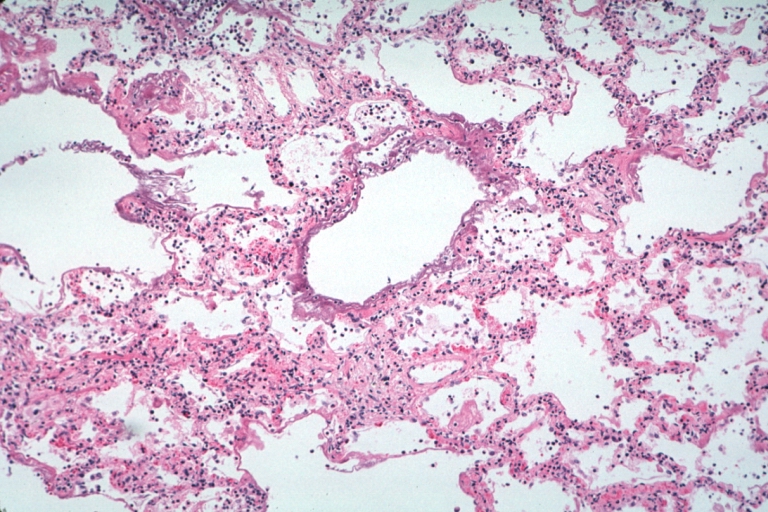
Lung: Necrotizing Bronchiolitis: Micro low mag H&E well shown necrotizing bronchiolitis and surrounding lesions of diffuse alveolar damage 19yo female with lupus erythematosus
-
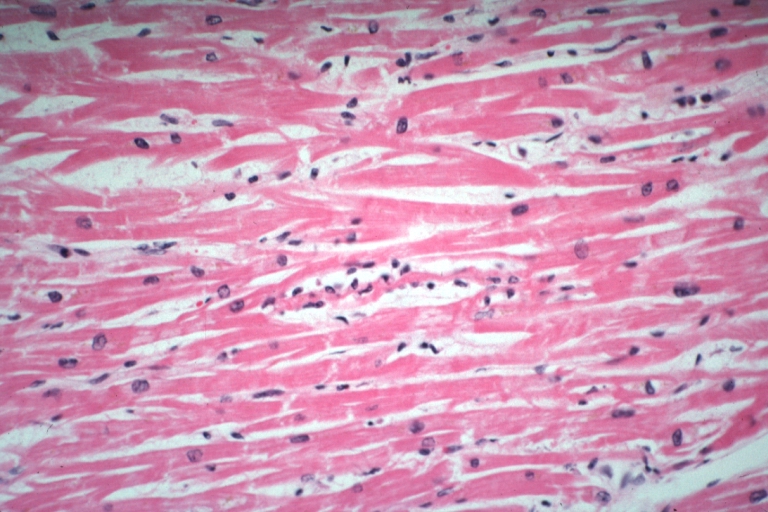
Myocarditis: Micro high mag H&E focal myofiber necrosis typical for this diagnosis but this is case of lupus erythematosus with Libman Sacks lesion and brain emboli and heart emboli did she also have viral myocarditis? This lesion is typical for the diagnosis
-
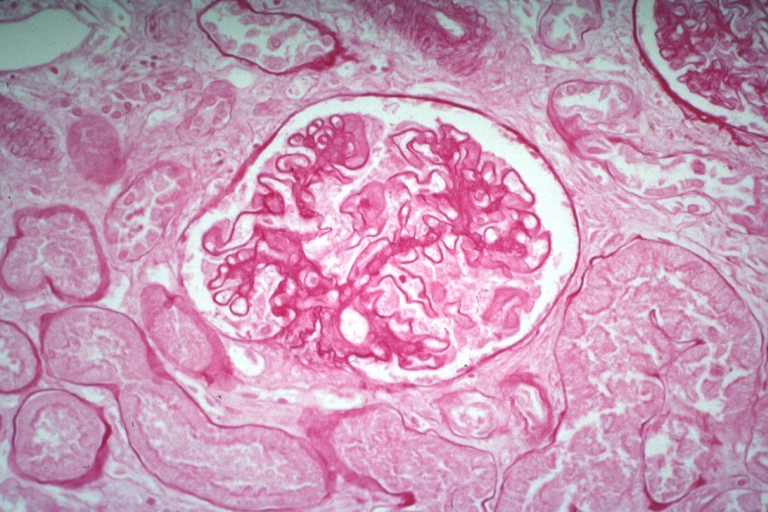
Kidney: Lupus Erythematosus: Micro high mag PAS stain thickened mesangium and capillary basement membranes 19yo female with renal failure and proved lupus
-
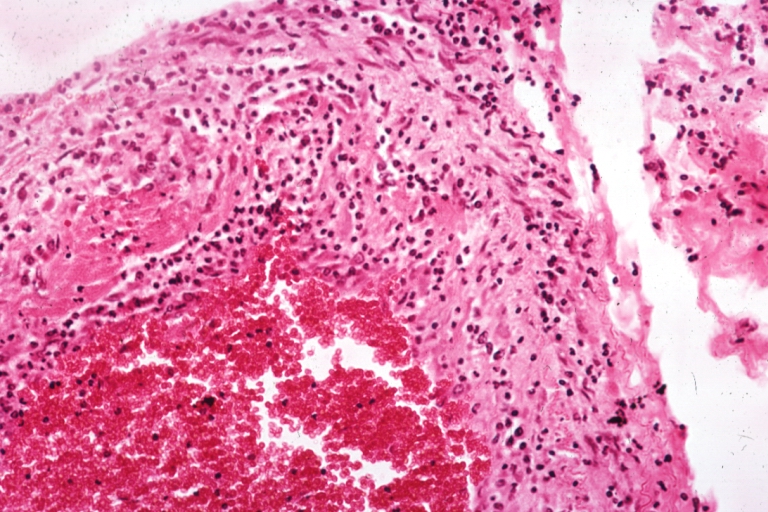
Artery: Arteritis in Lupus Erythematosus: Micro med mag H&E. A good example of vasculitis
-
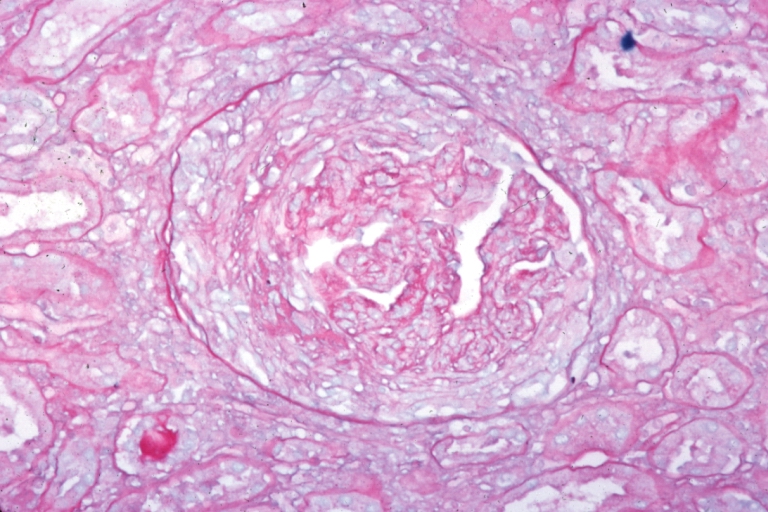
Kidney: Lupus Erythematosus: Micro high mag PASH typical glomerulonephritis lesion with crescent
-
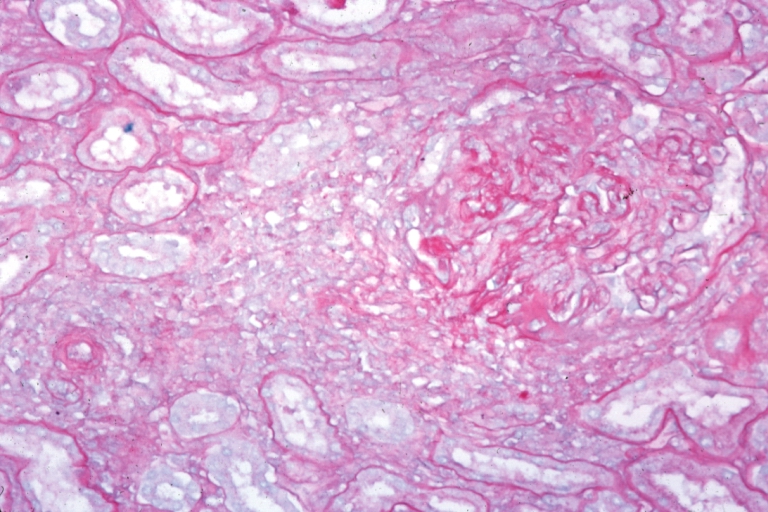
Kidney: Lupus Erythematosus: Micro med mag PASH glomerulonephritis
-
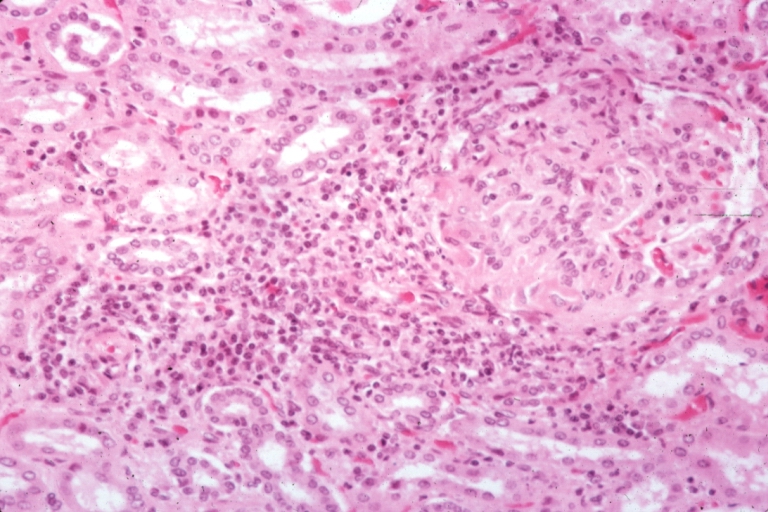
Kidney: Lupus Erythematosus: Micro med mag H&E typical glomerulonephritis lesion
-
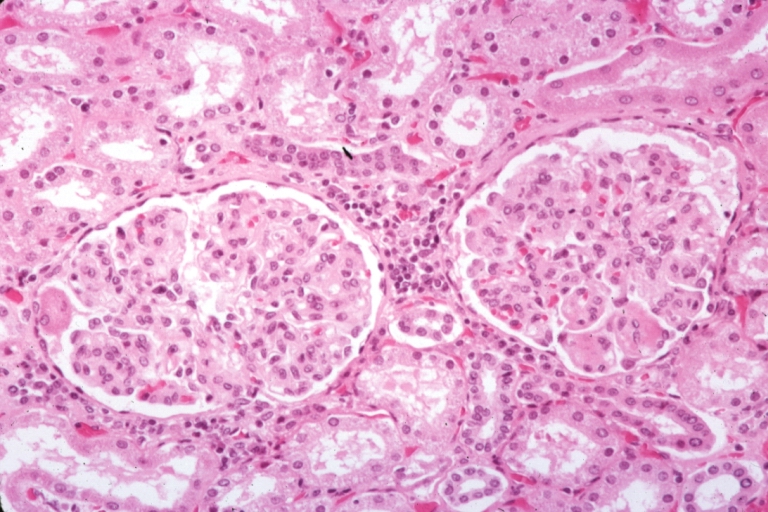
Kidney: Lupus Erythematosus: Micro med mag H&E two glomeruli showing lobular glomerulonephritis lesion
-
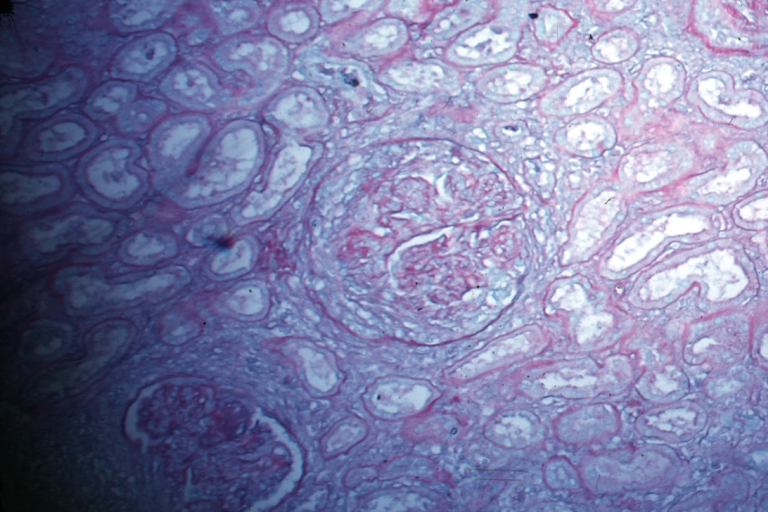
Kidney: Lupus Erythematosus: Micro med mag PASH typical chronic glomerulonephritis lesion with crescent
-
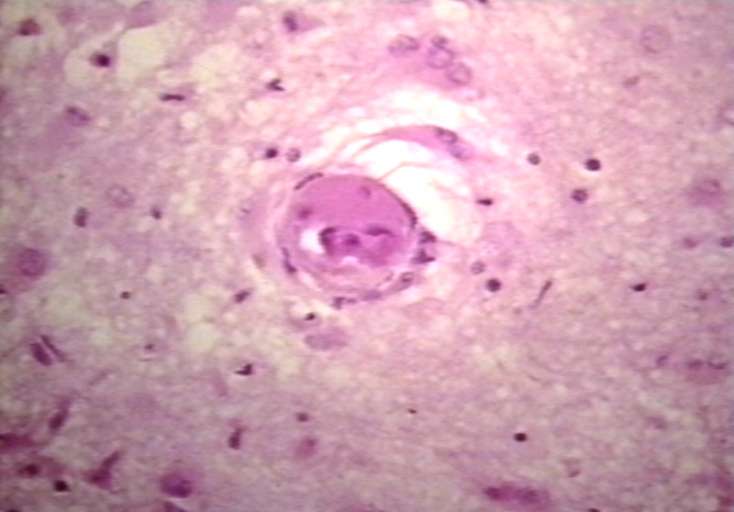
Vessel: lupus, systemic erythematosus; Thrombus in capillary
-
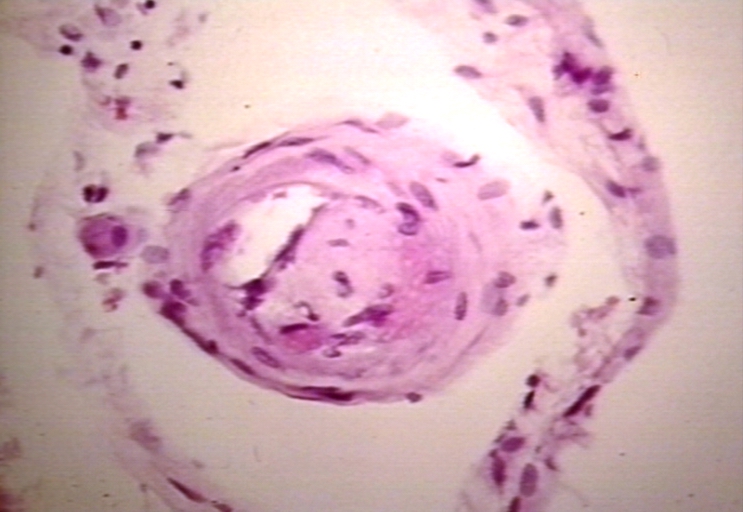
Vessel: lupus, systemic erythematosus; Thrombus in capillary
-
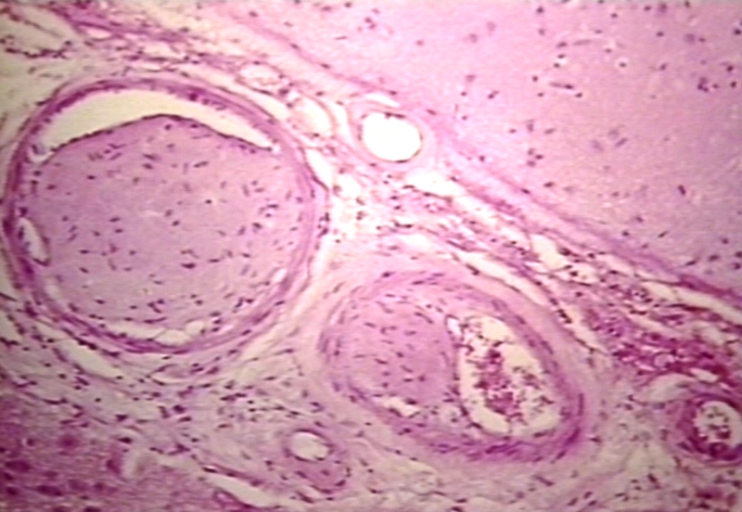
Vessel: lupus, systemic erythematosus; Thrombus in arteriole and vein
-
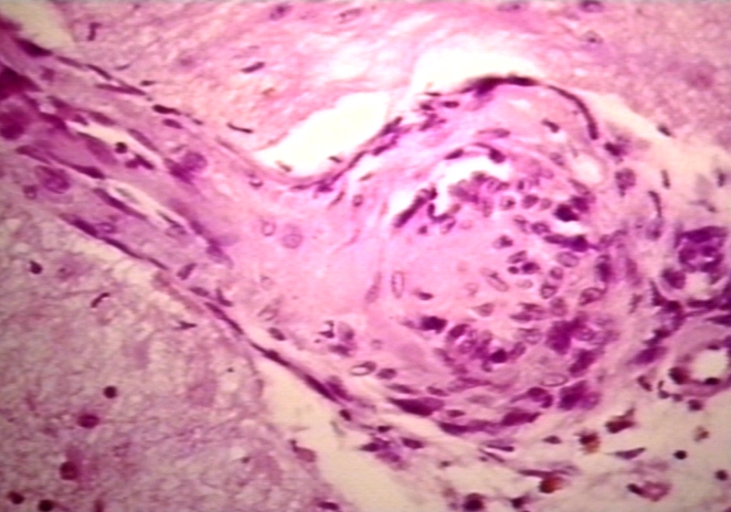
Vessel: lupus, systemic erythematosus; Thrombus in pial vessel
-
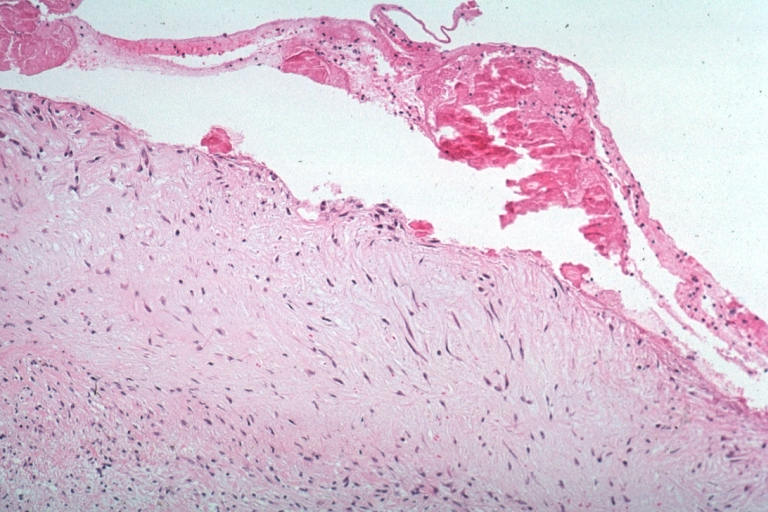
Lupus Erythematosus Libman Sacks Endocarditis: Micro high mag H&E atrial surface of mitral valve with small fibrin thrombus representing Libman Sacks lesion 10yo female
































Videos
{{#ev:youtube|Tw07BFaDEo0}}
References
- ↑ University of South Carolina School of Medicine lecture notes, Immunology, Hypersensitivity reactions. General discussion of hypersensitivity, not specific to SLE.
- ↑ Anisur Rahman and David A. Isenberg (February 28, 2008). "Review Article: Systemic Lupus Erythematosus". N Engl J Med. 358 (9): 929–939. doi:10.1056/NEJMra071297. PMID 18305268.
- ↑ Mary K. Crow (February 28, 2008). "Collaboration, Genetic Associations, and Lupus Erythematosus". N Engl J Med. 358 (9): 956–961. doi:10.1056/NEJMe0800096. PMID 18204099.
- ↑ Geoffrey Hom, Robert R. Graham, Barmak Modrek; et al. (February 28, 2008). "Association of Systemic Lupus Erythematosus with C8orf13–BLK and ITGAM–ITGAX". N Engl J Med. 358 (9): 900–909. doi:10.1056/NEJMoa0707865. PMID 18204098.
- ↑ Gaipl, U S; Kuhn, A; Sheriff, A; Munoz, L E; Franz, S; Voll, R E; Kalden, J R; Herrmann, M (2006). "Clearance of apoptotic cells in human SLE". Current directions in autoimmunity. 9: 173–87. PMID: 1639466 Abstract (full text requires registration).
- ↑ Poole BD, Schneider RI, Guthridge JM; et al. (2009). "Early targets of nuclear RNP humoral autoimmunity in human systemic lupus erythematosus". Arthritis Rheum. 60 (3): 848–859. doi:10.1002/art.24306. PMID 19248110. Unknown parameter
|month=ignored (help) - ↑ Pan HF, Wu GC, Li WP, Li XP, Ye DQ (2009). "High Mobility Group Box 1: a potential therapeutic target for systemic lupus erythematosus". Mol. Biol. Rep. doi:10.1007/s11033-009-9485-7. PMID 19247800. Unknown parameter
|month=ignored (help)