Romiplostim
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Aparna Vuppala, M.B.B.S. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Romiplostim is a Thrombopoietin Receptor Agonist that is FDA approved for the treatment of thrombocytopenia in patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins or splenectomy. Common adverse reactions include abdominal pain, diarrhea, indigestion, nausea, arthralgia, backache, myalgia, pain in limb, dizziness, headache, insomnia, paresthesia, epistaxis, upper respiratory infection, fatigue.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
- Romiplostim is indicated for the treatment of thrombocytopenia in patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins or splenectomy.
Limitations of use:
- Romiplostim is not indicated for the treatment of thrombocytopenia due to myelodysplastic syndrome (MDS) or any cause of thrombocytopenia other than chronic ITP.
- Romiplostim should be used only in patients with ITP whose degree of thrombocytopenia and clinical condition increases the risk for bleeding .
- Romiplostim should not be used in an attempt to normalize platelet counts .
Recommended Dosage Regimen
- Use the lowest dose of Romiplostim to achieve and maintain a platelet count ≥ 50 x 109/L as necessary to reduce the risk for bleeding. Administer Romiplostim as a weekly subcutaneous injection with dose adjustments based upon the platelet count response.
- The prescribed Romiplostim dose may consist of a very small volume (eg, 0.15 mL). Administer Romiplostim only with a syringe that contains 0.01 mL graduations.
Initial Dose
- The initial dose for Romiplostim is 1 mcg/kg based on actual body weight.
Dose Adjustments
- Use the actual body weight at initiation of therapy, then adjust the weekly dose of Romiplostim by increments of 1 mcg/kg until the patient achieves a platelet count ≥ 50 x 109/L as necessary to reduce the risk for bleeding; do not exceed a maximum weekly dose of 10 mcg/kg. In clinical studies, most patients who responded to Romiplostim achieved and maintained platelet counts ≥ 50 x 109/L with a median dose of 2 mcg/kg.
- During Romiplostim therapy, assess CBC, including platelet counts, weekly until a stable platelet count (≥ 50 x 109/L for at least 4 weeks without dose adjustment) has been achieved. Obtain CBC, including platelet counts, monthly thereafter.
- Adjust the dose as follows:
- If the platelet count is < 50 x 109/L, increase the dose by 1 mcg/kg.
- If platelet count is > 200 x 109/L for 2 consecutive weeks, reduce the dose by 1 mcg/kg.
- If platelet count is > 400 x 109/L, do not dose. Continue to assess the platelet count weekly. After the platelet count has fallen to < 200 x 109/L, resume Romiplostim at a dose reduced by 1 mcg/kg.
Discontinuation
- Discontinue Romiplostim if the platelet count does not increase to a level sufficient to avoid clinically important bleeding after 4 weeks of Romiplostim therapy at the maximum weekly dose of 10 mcg/kg . Obtain CBCs, including platelet counts, weekly for at least 2 weeks following discontinuation of Romiplostim .
Preparation and Administration
- To mitigate against medication errors (both overdose and underdose), ensure that these preparation and administration instructions are followed.
- Calculate the dose and reconstitute with the correct volume of sterile water for injection. Withdraw the appropriate volume of the calculated dose from the vial. Only administer subcutaneously .
- Romiplostim is supplied in single-use vials as a sterile, preservative-free, white lyophilized powder that must be reconstituted as outlined in Table 1 and administered using a syringe with 0.01 mL graduations. Using aseptic technique, reconstitute Romiplostim with preservative-free Sterile Water for Injection, USP as described in Table 1. Do not use bacteriostatic water for injection.
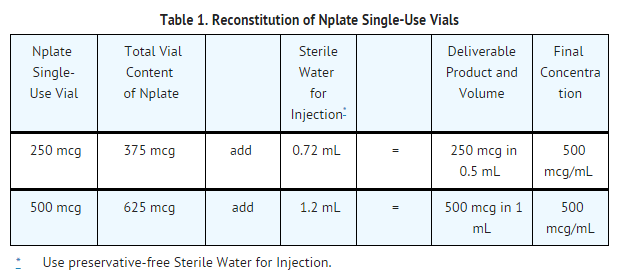
This image is provided by the National Library of Medicine.

- Gently swirl and invert the vial to reconstitute. Avoid excess or vigorous agitation: DO NOT SHAKE.Generally, dissolution of Romiplostim takes less than 2 minutes. The reconstituted Romiplostim solution should be clear and colorless. Visually inspect the reconstituted solution for particulate matter and/or discoloration. Do not administer Romiplostim if particulate matter and/or discoloration is observed.
- Reconstituted Romiplostim can be kept at room temperature (25°C/77°F) or refrigerated at 2° to 8°C (36° to 46°F) for up to 24 hours prior to administration. Protect the reconstituted product from light.
- To determine the injection volume to be administered, first identify the patient’s total dose in micrograms (mcg) using the dosing information . For example, a 75 kg patient initiating therapy at 1 mcg/kg will begin with a dose of 75 mcg. Next, calculate the volume of Romiplostim solution that is given to the patient by dividing the microgram dose by the concentration of the reconstituted Romiplostim solution (500 mcg/mL). For this patient example, the 75 mcg dose is divided by 500 mcg/mL, resulting in an injection volume of 0.15 mL.
- As the injection volume may be very small, use a syringe with graduations to 0.01 mL. Verify that the syringe contains the correct dosage.
- Discard any unused portion. Do not pool unused portions from the vials. Do not administer more than one dose from a vial.
Use of Romiplostim With Concomitant Medical ITP Therapies
- Romiplostim may be used with other medical ITP therapies, such as corticosteroids, danazol, azathioprine, intravenous immunoglobulin (IVIG), and anti-D immunoglobulin. If the patient’s platelet count is ≥ 50 x 109/L, medical ITP therapies may be reduced or discontinued
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Romiplostim in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Romiplostim in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Romiplostim in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Romiplostim in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Romiplostim in pediatric patients.
Contraindications
- None
Warnings
Risk of Progression of Myelodysplastic Syndromes to Acute Myelogenous Leukemia
- Progression from myelodysplastic syndromes (MDS) to acute myelogenous leukemia (AML) has been observed in clinical trials with Romiplostim. A randomized, double-blind, placebo-controlled trial enrolling patients with severe thrombocytopenia and International Prognostic Scoring System (IPSS) low or intermediate-1 risk MDS was terminated due to more cases of AML observed in the Romiplostim treatment arm. At the time of an interim analysis, among 219 MDS patients randomized 2:1 to treatment with Romiplostim or placebo (147 Romiplostim: 72 placebo), 11 patients showed progression to AML, including nine on the Romiplostim arm versus two on the placebo arm. In addition, in peripheral blood counts, the percentage of circulating myeloblasts increased to greater than 10% in 28 patients, 25 of whom were in the romiplostim treatment arm. Of the 28 patients who had an increase in circulating myeloblasts to greater than 10%, eight of these patients were diagnosed to have AML and 20 patients had not progressed to AML. In four patients, increased peripheral blood blast cell counts decreased to baseline after discontinuation of Romiplostim. In a single-arm trial of Romiplostim given to 72 patients with thrombocytopenia related to MDS, eight (11%) patients were reported as having possible disease progression, and three patients had confirmation of AML during follow-up. In addition, in three patients, increased peripheral blood blast cell counts decreased to baseline after discontinuation of Romiplostim.
- Romiplostim is not indicated for the treatment of thrombocytopenia due to MDS or any cause of thrombocytopenia other than chronic ITP.
Thrombotic/Thromboembolic Complications
- Thrombotic/thromboembolic complications may result from increases in platelet counts with Romiplostim use. Portal vein thrombosis has been reported in patients with chronic liver disease receiving Romiplostim. Romiplostim should be used with caution in patients with ITP and chronic liver disease.
- To minimize the risk for thrombotic/thromboembolic complications, do not use Romiplostim in an attempt to normalize platelet counts. Follow the dose adjustment guidelines to achieve and maintain a platelet count of ≥ 50 x 109/L .
Bone Marrow Reticulin Formation and Risk for Bone Marrow Fibrosis
- Romiplostim administration may increase the risk for development or progression of reticulin fiber formation within the bone marrow. This formation may improve upon discontinuation of Romiplostim. In a clinical trial, one patient with ITP and hemolytic anemia developed marrow fibrosis with collagen during Romiplostim therapy. Clinical trials are in progress to assess the risk of bone marrow fibrosis and clinical consequences with cytopenias.
- If new or worsening morphological abnormalities or cytopenia(s) occur, consider a bone marrow biopsy to include staining for fibrosis .
Worsened Thrombocytopenia after Cessation of Romiplostim
- In clinical studies of patients with chronic ITP who had Romiplostim discontinued, four of 57 patients developed thrombocytopenia of greater severity than was present prior to Romiplostim therapy. This worsened thrombocytopenia resolved within 14 days. Following discontinuation of Romiplostim, obtain weekly CBCs, including platelet counts, for at least 2 weeks and consider alternative treatments for worsening thrombocytopenia, according to current treatment guidelines .
Lack or Loss ofResponse to Romiplostim
- Hyporesponsiveness or failure to maintain a platelet response with Romiplostim should prompt a search for causative factors, including neutralizing antibodies to Romiplostim. To detect antibody formation, submit blood samples to Amgen (1-800-772-6436). Amgen will assay these samples for antibodies to Romiplostim and thrombopoietin (TPO). Discontinue Romiplostim if the platelet count does not increase to a level sufficient to avoid clinically important bleeding after 4 weeks at the highest weekly dose of 10 mcg/kg.
Adverse Reactions
Clinical Trials Experience
- Serious adverse reactions associated with Romiplostim in ITP clinical studies were bone marrow reticulin deposition and worsening thrombocytopenia after Romiplostim discontinuation
- The data described below reflect Romiplostim exposure to 271 patients with chronic ITP, aged 18 to 88, of whom 62% were female. Romiplostim was studied in two randomized, placebo-controlled, double-blind studies that were identical in design, with the exception that Study 1 evaluated nonsplenectomized patients with ITP and Study 2 evaluated splenectomized patients with ITP. Data are also reported from an open-label, single-arm study in which patients received Romiplostim over an extended period of time. Overall, Romiplostim was administered to 114 patients for at least 52 weeks and 53 patients for at least 96 weeks.
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
- In the placebo-controlled studies, headache was the most commonly reported adverse drug reaction, occurring in 35% of patients receiving Romiplostim and 32% of patients receiving placebo. Headaches were usually of mild or moderate severity. Table 2 presents adverse drug reactions from Studies 1 and 2 with a ≥ 5% higher patient incidence in Romiplostim versus placebo. The majority of these adverse drug reactions were mild to moderate in severity.
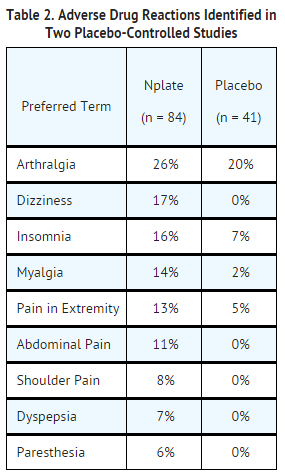
This image is provided by the National Library of Medicine.

- Among 142 patients with chronic ITP who received Romiplostim in the single-arm extension study, the incidence rates of the adverse reactions occurred in a pattern similar to those reported in the placebo-controlled clinical studies.
Postmarketing Experience
- The following adverse reactions have been identified during post approval use of Romiplostim. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immunogenicity
- As with all therapeutic proteins, patients may develop antibodies to the therapeutic protein. Patients were screened for immunogenicity to romiplostim using a BIAcore-based biosensor immunoassay. This assay is capable of detecting both high- and low-affinity binding antibodies that bind to romiplostim and cross-react with TPO. The samples from patients that tested positive for binding antibodies were further evaluated for neutralizing capacity using a cell-based bioassay.
- In clinical studies, the incidence of preexisting antibodies to romiplostim was 8% (43/537) and the incidence of binding antibody development during Romiplostim treatment was 6% (31/537). The incidence of preexisting antibodies to endogenous TPO was 5% (29/537) and the incidence of binding antibody development to endogenous TPO during Romiplostim treatment was 4% (21/537). Of the patients with positive binding antibodies that developed to romiplostim or to TPO, two (0.4%) patients had neutralizing activity to romiplostim and none had neutralizing activity to TPO. No correlation was observed between antibody activity and clinical effectiveness or safety.
- Immunogenicity assay results are highly dependent on the sensitivity and specificity of the assay used in detection and may be influenced by several factors, including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to romiplostim with the incidence of antibodies to other products may be misleading.
Drug Interactions
- No formal drug interaction studies of Romiplostim have been performed.
Use in Specific Populations
Pregnancy
- There are no adequate and well-controlled studies of Romiplostim use in pregnant women. In animal reproduction and developmental toxicity studies, romiplostim crossed the placenta, and adverse fetal effects included thrombocytosis, postimplantation loss, and an increase in pup mortality. Romiplostim should be used during pregnancy only if the potential benefit to the mother justifies the potential risk to the fetus.
- Pregnancy Registry: A pregnancy registry has been established to collect information about the effects of Romiplostim use during pregnancy. Physicians are encouraged to register pregnant patients, or pregnant women may enroll themselves in the Romiplostim pregnancy registry by calling 1-800-77-AMGEN (1-800-772-6436).
- In rat and rabbit developmental toxicity studies, no evidence of fetal harm was observed at romiplostim doses up to 11 times (rats) and 82 times (rabbits) the maximum human dose (MHD) based on systemic exposure. In mice at doses 5 times the MHD, reductions in maternal body weight and increased postimplantation loss occurred.
- In a prenatal and postnatal development study in rats, at doses 11 times the MHD, there was an increase in perinatal pup mortality. Romiplostim crossed the placental barrier in rats and increased fetal platelet counts at clinically equivalent and higher doses.
Pregnancy Category (AUS):
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Romiplostim in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Romiplostim during labor and delivery.
Nursing Mothers
- It is not known whether Romiplostim is excreted in human milk; however, human IgG is excreted in human milk. Published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Romiplostim, a decision should be made whether to discontinue nursing or to discontinue Romiplostim, taking into account the importance of Romiplostim to the mother and the known benefits of nursing.
Pediatric Use
- The safety and effectiveness in pediatric patients (<18 years) have not been established.
Geriatic Use
- Of the 271 patients who received Romiplostim in ITP clinical studies, 55 (20%) were age 65 and over, and 27 (10%) were 75 and over. No overall differences in safety or efficacy have been observed between older and younger patients in the placebo-controlled studies, but greater sensitivity of some older individuals cannot be ruled out. In general, dose adjustment for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Gender
There is no FDA guidance on the use of Romiplostim with respect to specific gender populations.
Race
There is no FDA guidance on the use of Romiplostim with respect to specific racial populations.
Renal Impairment
- No clinical studies were conducted in patients with renal impairment. Use Romiplostim with caution in this population.
Hepatic Impairment
- No clinical studies were conducted in patients with hepatic impairment. Use Romiplostim with caution in this population.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Romiplostim in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Romiplostim in patients who are immunocompromised.
Administration and Monitoring
Administration
Monitoring
- Obtain CBCs, including platelet counts, weekly during the dose-adjustment phase of Romiplostim therapy and then monthly following establishment of a stable Romiplostim dose.
- Obtain CBCs, including platelet counts, weekly for at least 2 weeks following discontinuation of Romiplostim
IV Compatibility
There is limited information regarding IV Compatibility of Romiplostim in the drug label.
Overdosage
- Overdoses due to medication errors have been reported in patients receiving Romiplostim. In the event of overdose, platelet counts may increase excessively and result in thrombotic/thromboembolic complications. In this case, discontinue Romiplostim and monitor platelet counts. Reinitiate treatment with Romiplostim in accordance with dosing and administration recommendations
Pharmacology
Mechanism of Action
- Romiplostim increases platelet production through binding and activation of the TPO receptor, a mechanism analogous to endogenous TPO.
Structure
- Romiplostim, a member of the TPO mimetic class, is an Fc-peptide fusion protein (peptibody) that activates intracellular transcriptional pathways leading to increased platelet production via the TPO receptor (also known as cMpl). The peptibody molecule contains two identical single-chain subunits, each consisting of human immunoglobulin IgG1 Fc domain, covalently linked at the C-terminus to a peptide containing two thrombopoietin receptor-binding domains. Romiplostim has no amino acid sequence homology to endogenous TPO. Romiplostim is produced by recombinant DNA technology in Escherichia coli (E coli).
- Romiplostim is supplied as a sterile, preservative-free, lyophilized, solid white powder for subcutaneous injection. Two vial presentations are available, which contain a sufficient amount of active ingredient to provide either 250 mcg or 500 mcg of deliverable romiplostim, respectively. Each single-use 250 mcg vial of Romiplostim contains the following: 375 mcg romiplostim, 30 mg mannitol, 15 mg sucrose, 1.2 mg L-histidine, 0.03 mg polysorbate 20, and sufficient HCl to adjust the pH to a target of 5.0. Each single-use 500 mcg vial of Romiplostim contains the following: 625 mcg romiplostim, 50 mg mannitol, 25 mg sucrose, 1.9 mg L-histidine, 0.05 mg polysorbate 20, and sufficient HCl to adjust the pH to a target of 5.0
Pharmacodynamics
- In clinical studies, treatment with Romiplostim resulted in dose-dependent increases in platelet counts. After a single subcutaneous dose of 1 to 10 mcg/kg Romiplostim in patients with chronic ITP, the peak platelet count was 1.3 to 14.9 times greater than the baseline platelet count over a 2- to 3-week period. The platelet counts were above 50 x 109/L for seven out of eight patients with chronic ITP who received six weekly doses of Romiplostim at 1 mcg/kg.
Pharmacokinetics
- In the long-term extension study in patients with ITP receiving weekly treatment of Romiplostim subcutaneously, the pharmacokinetics of romiplostim over the dose range of 3 to 15 mcg/kg indicated that peak serum concentrations of romiplostim were observed about 7 to 50 hours post dose (median: 14 hours) with half-life values ranging from 1 to 34 days (median: 3.5 days). The serum concentrations varied among patients and did not correlate with the dose administered. The elimination of serum romiplostim is in part dependent on the TPO receptor on platelets. As a result, for a given dose, patients with high platelet counts are associated with low serum concentrations and vice versa. In another ITP clinical study, no accumulation in serum concentrations was observed (n = 4) after six weekly doses of Romiplostim (3 mcg/kg). The accumulation at higher doses of romiplostim is unknown.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment of Fertility
- The carcinogenic potential of romiplostim has not been evaluated. The mutagenic potential of romiplostim has not been evaluated. Romiplostim had no effect on the fertility of rats at doses up to 37 times the MHD based on systemic exposure.
Animal Toxicology and/or Pharmacology
- In a 4-week repeat-dose toxicity study in which rats were dosed subcutaneously three times per week, romiplostim caused extramedullary hematopoiesis, bone hyperostosis, and marrow fibrosis at clinically equivalent and higher doses. In this study, these findings were not observed in animals after a 4-week post treatment recovery period. Studies of long-term treatment with romiplostim in rats have not been conducted; therefore, it is not known if the fibrosis of the bone marrow is reversible in rats after long-term treatment.
Clinical Studies
Chronic ITP
- The safety and efficacy of Romiplostim were assessed in two double-blind, placebo-controlled clinical studies and in an open-label extension study.
Studies 1 and 2
- In Studies 1 and 2, patients with chronic ITP who had completed at least one prior treatment and had a platelet count of ≤ 30 x 109/L prior to study entry were randomized (2:1) to 24 weeks of Romiplostim (1 mcg/kg subcutaneous [SC]) or placebo. Prior ITP treatments in both study groups included corticosteroids, immunoglobulins, rituximab, cytotoxic therapies, danazol, and azathioprine. Patients already receiving ITP medical therapies at a constant dosing schedule were allowed to continue receiving these medical treatments throughout the studies. Rescue therapies (ie, corticosteroids, IVIG, platelet transfusions, and anti-D immunoglobulin) were permitted for bleeding, wet purpura, or if the patient was at immediate risk for hemorrhage. Patients received single weekly SC injections of Romiplostim, with individual dose adjustments to maintain platelet counts (50 x 109/L to 200 x 109/L).
- Study 1 evaluated patients who had not undergone a splenectomy. The patients had been diagnosed with ITP for approximately 2 years and had received a median of three prior ITP treatments. Overall, the median platelet count was 19 x 109/L at study entry. During the study, the median weekly Romiplostim dose was 2 mcg/kg (25th–75th percentile: 1–3 mcg/kg).
- Study 2 evaluated patients who had undergone a splenectomy. The patients had been diagnosed with ITP for approximately 8 years and had received a median of six prior ITP treatments. Overall, the median platelet count was 14 x 109/L at study entry. During the study, the median weekly Romiplostim dose was 3 mcg/kg (25th–75th percentile: 2–7 mcg/kg).
- Study 1 and 2 outcomes are shown in Table 3. A durable platelet response was the achievement of a weekly platelet count ≥ 50 x 109/L for any 6 of the last 8 weeks of the 24-week treatment period in the absence of rescue medication at any time. A transient platelet response was the achievement of any weekly platelet counts ≥ 50 x 109/L for any 4 weeks during the treatment period without a durable platelet response. An overall platelet response was the achievement of either a durable or a transient platelet response. Platelet responses were excluded for 8 weeks after receiving rescue medications.
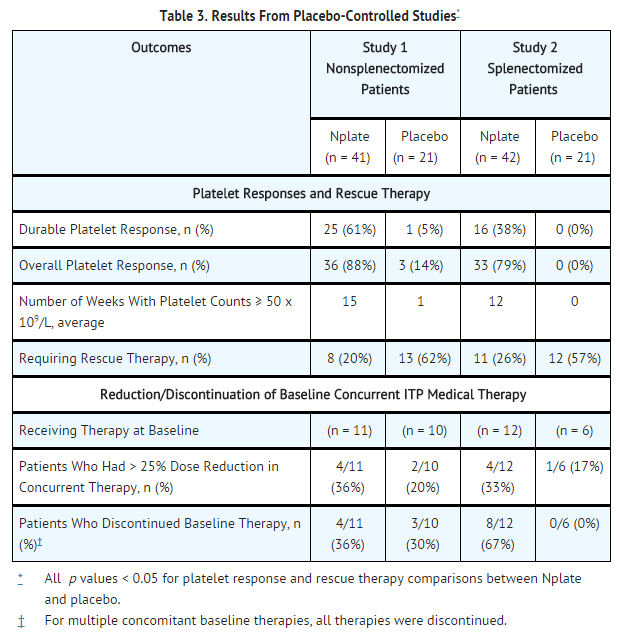
This image is provided by the National Library of Medicine.

- In Studies 1 and 2, nine patients reported a serious bleeding event [five (6%) Romiplostim, four (10%) placebo]. Bleeding events that were grade 2 severity or higher occurred in 15% of patients treated with Romiplostim and 34% of patients treated with placebo.
Extension Study
- Patients who had participated in either Study 1 or Study 2 were withdrawn from study medications. If platelet counts subsequently decreased to ≤ 50 x 109/L, the patients were allowed to receive Romiplostim in an open-label extension study with weekly dosing based on platelet counts. Following Romiplostim discontinuation in Studies 1 and 2, seven patients maintained platelet counts of ≥ 50 x 109/L. Among 100 patients who subsequently entered the extension study, platelet counts were increased and sustained regardless of whether they had received Romiplostim or placebo in the prior placebo-controlled studies. The majority of patients reached a median platelet count of 50 x 109/L after receiving one to three doses of Romiplostim, and these platelet counts were maintained throughout the remainder of the study with a median duration of Romiplostim treatment of 60 weeks and a maximum duration of 96 weeks.
How Supplied
- Romiplostim is supplied in single-use vials containing 250 mcg (NDC 55513-221-01) and 500 mcg (NDC 55513-222-01) deliverable romiplostim.
Storage
- Store Romiplostim vials in their carton to protect from light until time of use. Keep Romiplostim vials refrigerated at 2° to 8°C (36° to 46°F). Do not freeze.
Images
Drug Images
{{#ask: Page Name::Romiplostim |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Romiplostim |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- Prior to treatment, patients should fully understand the risks and benefits of Romiplostim. Inform patients that the risks associated with long-term administration of Romiplostim are unknown.
- Inform patients of the following risks and considerations for Romiplostim:
- Romiplostim therapy is administered to achieve and maintain a platelet count ≥ 50 x 109/L as necessary to reduce the risk for bleeding; Romiplostim is not used to normalize platelet counts.
- Following discontinuation of Romiplostim, thrombocytopenia and risk of bleeding may develop that is worse than that experienced prior to the Romiplostim therapy.
- Romiplostim therapy may increase the risk of reticulin fiber formation within the bone marrow. This formation may improve upon discontinuation. Detection of peripheral blood cell abnormalities may necessitate a bone marrow examination.
- Too much Romiplostim may result in excessive platelet counts and a risk for thrombotic/thromboembolic complications.
- Romiplostim stimulates certain bone marrow cells to make platelets and increases the risk of progression to acute myelogenous leukemia in patients with myelodysplastic syndromes.
- Platelet counts and CBCs must be performed weekly until a stable Romiplostim dose has been achieved; thereafter, platelet counts and CBCs, must be performed monthly while taking Romiplostim.
- Patients must be closely monitored with weekly platelet counts and CBCs for at least 2 weeks following Romiplostim discontinuation.
- Even with Romiplostim therapy, patients should continue to avoid situations or medications that may increase the risk for bleeding.
Precautions with Alcohol
- Alcohol-Romiplostim interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- Nplate
Look-Alike Drug Names
- romiPLOStim - romiDEPsin
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Label Page=Romiplostim |Label Name=Romiplostim05.png
}}
{{#subobject:
|Label Page=Romiplostim |Label Name=Romiplostim06.png
}}
{{#subobject:
|Label Page=Romiplostim |Label Name=Romiplostim07.png
}}