Pheochromocytoma
| Pheochromocytoma | |
 | |
|---|---|
| Micrograph of a pheochromocytoma. | |
| ICD-10 | C74.1 |
| ICD-9 | 255.6 |
| ICD-O: | Template:ICDO |
| OMIM | 171300 |
| DiseasesDB | 9912 |
| MeSH | D010673 |
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
Differentiating Pheochromocytoma from other Diseases
Causes of Pheochromocytoma
History & Symptoms
Tumor Location
In adults, 90% tumors are located unilaterally and are solitary, and 10% are located outside the adrenal gland. In children 50% are adrenal, while 25% are bilateral and 25% are extraadrenal. The common extradrenal locations are the abdomen, thorax and urinary bladder.
Diagnosis
_histopathology.jpg)


The diagnosis can be established by measuring catecholamines and metanephrines in plasma or through a 24-hour urine collection. Care should be taken to rule out other causes of adrenergic (adrenalin-like) excess like hypoglycemia, stress, exercise, and drugs affecting the catecholamines like stimulants, methyldopa, dopamine agonists, or ganglion blocking antihypertensives. Various foodstuffs (e.g. vanilla ice cream) can also affect the levels of urinary metanephrine and VMA (vanillyl mandelic acid). Imaging by computed tomography or a T2 weighted MRI of the head, neck, and chest, and abdomen can help localize the tumor. Tumors can also be located using Iodine-131 meta-iodobenzylguanidine (I131 MIBG) imaging.
One diagnostic test used in the past for a pheochromocytoma is to administer clonidine, a centrally-acting alpha-2 agonist used to treat high blood pressure. Clonidine mimics catecholamines in the brain, causing it to reduce the activity of the sympathetic nerves controlling the adrenal medulla. A healthy adrenal medulla will respond to the Clonidine suppression test by reducing catecholamine production; the lack of a response is evidence of pheochromocytoma.
Another test is for the clinician to press gently on the adrenal gland. A pheochromocytoma will often release a burst of catecholamines, with the associated signs and symptoms quickly following. This method is not recommended because of possible complications arising from a potentially massive release of catecholamines.
Pheochromocytomas occur most often during young-adult to mid-adult life. Less than 10% of pheochromocytomas are malignant (cancerous), bilateral or pediatric.
These tumors can form a pattern with other endocrine gland cancers which is labeled multiple endocrine neoplasia (MEN). Pheochromocytoma may occur in patients with MEN 2 and MEN 3. VHL (Von Hippel Lindau) patients may also develop these tumors.
Patients experiencing symptoms associated with pheochromocytoma should be aware that it is rare. However, it often goes undiagnosed until autopsy; therefore patients might wisely choose to take steps to provide a physician with important clues, such as recording whether blood pressure changes significantly during episodes of apparent anxiety.
Medical Therapy
Surgical resection of the tumor is the treatment of first choice. Given the complexity of perioperative management, and the potential for catastrophic intra and postoperative complications, such surgery should be performed only at centers experienced in the area. In addition to the surgical expertise that such centers can provide, they will also have the necessary endocrine and anesthesia resources as well. It may also be nescessary to carry out adrenalectomy, a complete surgical removal of the affected adrenal gland(s).
Either surgical option requires prior treatment with both the non-specific alpha adrenoceptor blocker Phenoxybenzamine to counteract hypertension and the beta-1 adrenoceptor antagonist Atenolol to reduce cardiac output. Given before surgery, these can also block the effect of a sudden release of adrenaline during tumour removal, which would otherwise endanger the anaethetised patient.
Historical perspective
In 1886, Fränkel made the first description of a patient with pheochromocytoma, however the term was first coined by Pick, a pathologist, in 1912. In 1926, Roux (in Switzerland) and Mayo (in U.S.A.) were the first surgeons to remove pheochromocytomas.
Jaroszewski DE, Tessier DJ, Schlinkert RT, et al. Laparoscopic adrenalectomy for pheochromocytoma. Mayo Clin Proc. 2003; 78: 1501-1504.
Additional images
-
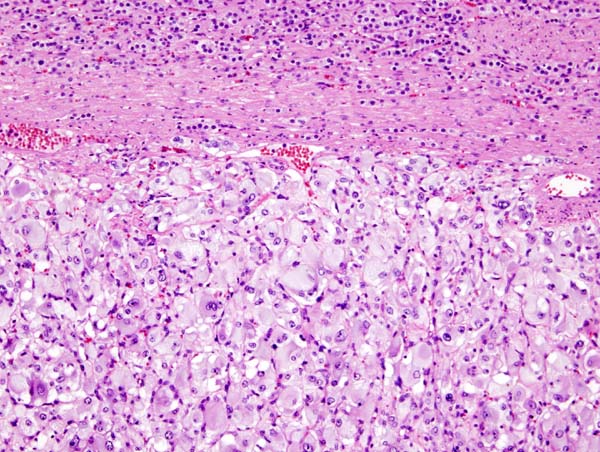
Micrograph of pheochromocytoma.
-
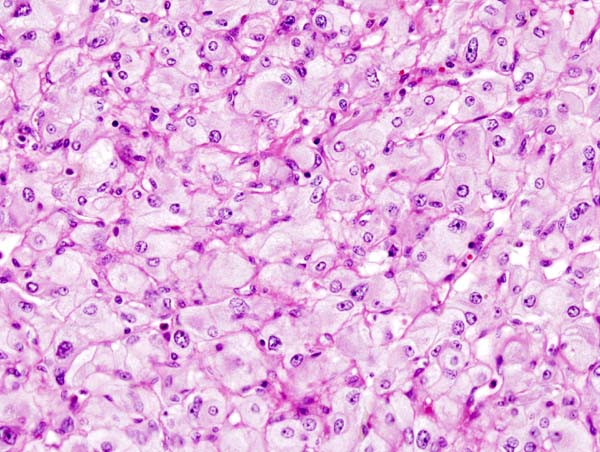
Micrograph of pheochromocytoma.
-
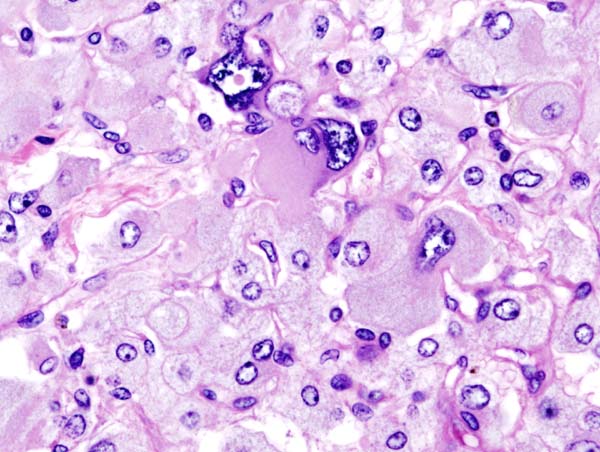
Micrograph of pheochromocytoma.
-
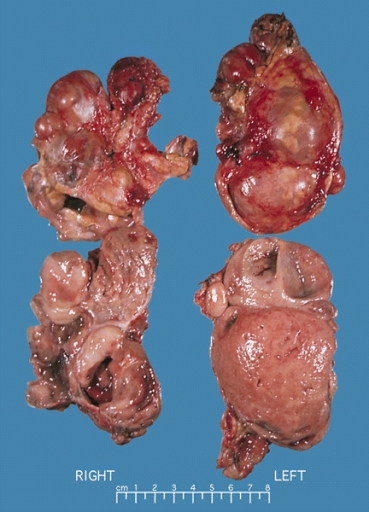
Bilateral pheochromocytoma in MEN2. Gross image.
-
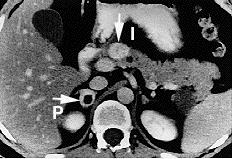
Pheochromocytoma. CT abdomen.
-
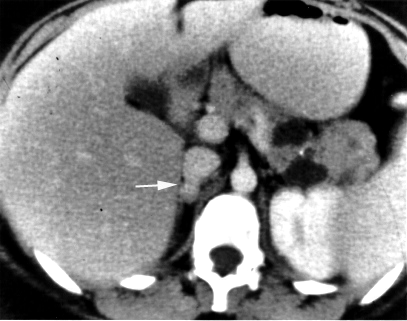
Pheochromocytoma. CT abdomen.
_histopathology.jpg)
_histopathology.jpg)



References
External links
- Pheochromocytoma clinical trial currently recruiting patients
- Template:MedlinePlusOverview
- overview from National Cancer Institute
- Pheochromocytoma Research Support Organization at pressor.org
- Pheochromocytoma Support Worldwide at pub1.ezboard.com
- pheochromocytoma.org
Template:Epithelial neoplasms Template:SIB de:Phäochromozytom it:Feocromocitoma he:פאוכרומוציטומה nl:Feochromocytoom sv:Feokromocytom