Pheochromocytoma
| Pheochromocytoma | |
 | |
|---|---|
| Micrograph of a pheochromocytoma. | |
| ICD-10 | C74.1 |
| ICD-9 | 255.6 |
| ICD-O: | Template:ICDO |
| OMIM | 171300 |
| DiseasesDB | 9912 |
| MedlinePlus | 000340 |
| MeSH | D010673 |
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
A pheochromocytoma (phaeochromocytoma outside of the US) is a neuroendocrine tumor of the medulla of the adrenal glands (originating in the chromaffin cells) or extra-adrenal chromaffin tissue which failed to involute after birth,[1] which secretes excessive amounts of catecholamines, usually epinephrine and norepinephrine. Extra-adrenal paragangliomas (often described as extra-adrenal pheochromocytomas) are closely related, though less common, tumors that originate in the ganglia of the sympathetic nervous system and are named based upon the primary anatomical site of origin.
Traditionally it is known as the "10% tumor":
- bilateral disease is present in approximately 10% of patients
- approximately 10% of tumours are malignant
- approximately 10% are located in chromaffin tissue outside of the adrenal gland
- Approximately 10% arise in childhood
- Approximately 10% are familial
- Approximately 10% recur after being resected
- Approximately 10% patients do not have hypertension (Campbell's Urology)
Differentiating Pheochromocytoma from other Diseases
The differential diagnosis of pheochromocytoma includes:
- Anxiety disorders
- Carcinoid tumor
- Paragangliomas
- Essential hypertension
- Hyperthyroidism
- Insulinoma
- Paroxysmal supraventricular tachycardia
- Renovascular hypertension
Causes of Pheochromocytoma
Up to 25% of pheochromocytomas may be familial. Mutations of the genes VHL, RET, NF1, SDHB and SDHD are all known to cause familial pheochromocytoma/extra-adrenal paraganglioma.
Pheochromocytoma is a tumor of the multiple endocrine neoplasia syndrome, type IIA (also known as MEN IIA) and type IIB MEN IIB. The other component neoplasms of that syndrome include parathyroid adenomas, and medullary thyroid cancer. Mutations in the autosomal RET proto-oncogene drives these malignancies [2].
Common mutations in the RET oncogene may also account for medullary sponge kidney as well. [3]
Pheochromocytoma linked to MEN II can be caused by RET oncogene mutations. Both syndromes are characterized by pheochromocytoma as well as thyroid cancer (thyroid medullary carcinoma). MEN IIA also presents with hyperparathyroidism, while MEN IIB also presents with mucosal neuroma. It is now postulated that Lincoln suffered from MEN IIB, rather than Marfan's syndrome as previously thought, though this is uncertain.
History & Symptoms
The signs and symptoms of a pheochromocytoma are those of sympathetic nervous system hyperactivity, including:
- Elevated heart rate
- Elevated blood pressure, including paroxysmal (sporadic, episodic) high blood pressure, which sometimes can be more difficult to detect; another clue to the presence of pheochromocytoma is orthostatic hypotension (a fall in systolic blood pressure greater than 20 mmHg or a fall in diastolic blood pressure greater than 10 mmHg on making the patient stand)
- Palpitations
- Anxiety often resembling that of a panic attack
- Diaphoresis
- Headaches
- Pallor
- Weight loss
- Localized amyloid deposits found microscopically
- Elevated blood glucose level (due primarily to catecholamine stimulation of lipolysis (breakdown of stored fat) leading to high levels of free fatty acids and the subsequent inhibition of glucose uptake by muscle cells. Further, stimulation of beta-adrenergic receptors leads to glycogenolysis and gluconeogenesis and thus elevation of blood glucose levels).
A pheochromocytoma can also cause resistant arterial hypertension. A pheochromocytoma can be fatal if it causes malignant hypertension, or severely high blood pressure. This hypertension is not well controlled with standard blood pressure medications.
Not all patients experience all of the signs and symptoms listed. The most common presentation is headache, excessive sweating, and increased heart rate, with the attack subsiding in less than one hour.
Tumors may grow very large, but most are smaller than 10 cm.
Tumor Location
In adults, 90% tumors are located unilaterally and are solitary, and 10% are located outside the adrenal gland. In children 50% are adrenal, while 25% are bilateral and 25% are extraadrenal. The common extradrenal locations are the abdomen, thorax and urinary bladder.
Diagnosis
_histopathology.jpg)


The diagnosis can be established by measuring catecholamines and metanephrines in plasma or through a 24-hour urine collection. Care should be taken to rule out other causes of adrenergic (adrenalin-like) excess like hypoglycemia, stress, exercise, and drugs affecting the catecholamines like stimulants, methyldopa, dopamine agonists, or ganglion blocking antihypertensives. Various foodstuffs (e.g. vanilla ice cream) can also affect the levels of urinary metanephrine and VMA (vanillyl mandelic acid). Imaging by computed tomography or a T2 weighted MRI of the head, neck, and chest, and abdomen can help localize the tumor. Tumors can also be located using Iodine-131 meta-iodobenzylguanidine (I131 MIBG) imaging.
One diagnostic test used in the past for a pheochromocytoma is to administer clonidine, a centrally-acting alpha-2 agonist used to treat high blood pressure. Clonidine mimics catecholamines in the brain, causing it to reduce the activity of the sympathetic nerves controlling the adrenal medulla. A healthy adrenal medulla will respond to the Clonidine suppression test by reducing catecholamine production; the lack of a response is evidence of pheochromocytoma.
Another test is for the clinician to press gently on the adrenal gland. A pheochromocytoma will often release a burst of catecholamines, with the associated signs and symptoms quickly following. This method is not recommended because of possible complications arising from a potentially massive release of catecholamines.
Pheochromocytomas occur most often during young-adult to mid-adult life. Less than 10% of pheochromocytomas are malignant (cancerous), bilateral or pediatric.
These tumors can form a pattern with other endocrine gland cancers which is labeled multiple endocrine neoplasia (MEN). Pheochromocytoma may occur in patients with MEN 2 and MEN 3. VHL (Von Hippel Lindau) patients may also develop these tumors.
Patients experiencing symptoms associated with pheochromocytoma should be aware that it is rare. However, it often goes undiagnosed until autopsy; therefore patients might wisely choose to take steps to provide a physician with important clues, such as recording whether blood pressure changes significantly during episodes of apparent anxiety.
Treatment
Surgical resection of the tumor is the treatment of first choice. Given the complexity of perioperative management, and the potential for catastrophic intra and postoperative complications, such surgery should be performed only at centers experienced in the area. In addition to the surgical expertise that such centers can provide, they will also have the necessary endocrine and anesthesia resources as well. It may also be nescessary to carry out adrenalectomy, a complete surgical removal of the affected adrenal gland(s).
Either surgical option requires prior treatment with both the non-specific alpha adrenoceptor blocker Phenoxybenzamine to counteract hypertension and the beta-1 adrenoceptor antagonist Atenolol to reduce cardiac output. Given before surgery, these can also block the effect of a sudden release of adrenaline during tumour removal, which would otherwise endanger the anaethetised patient.
Historical
In 1886, Fränkel made the first description of a patient with pheochromocytoma, however the term was first coined by Pick, a pathologist, in 1912. In 1926, Roux (in Switzerland) and Mayo (in U.S.A.) were the first surgeons to remove pheochromocytomas.
Jaroszewski DE, Tessier DJ, Schlinkert RT, et al. Laparoscopic adrenalectomy for pheochromocytoma. Mayo Clin Proc. 2003; 78: 1501-1504.
Additional images
-
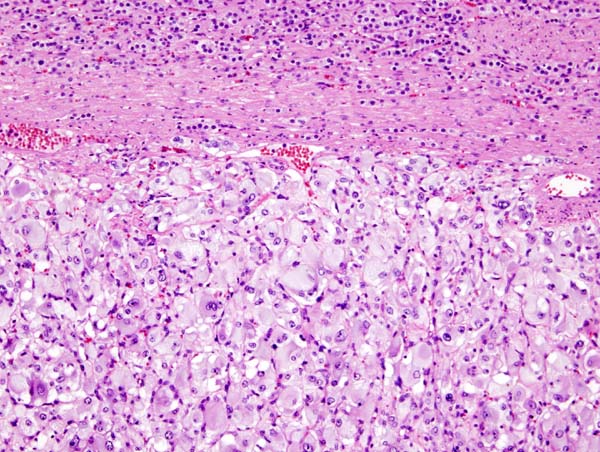
Micrograph of pheochromocytoma.
-
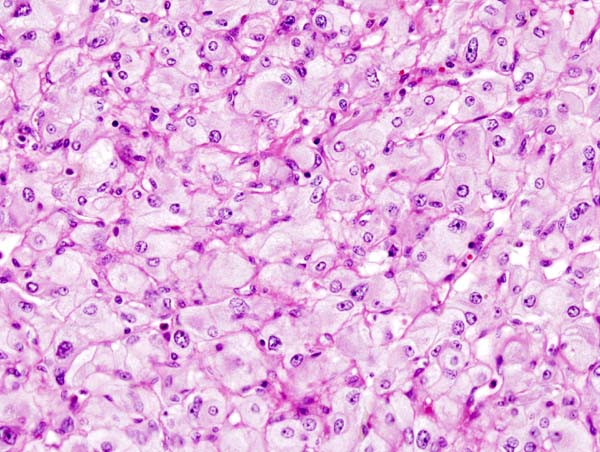
Micrograph of pheochromocytoma.
-
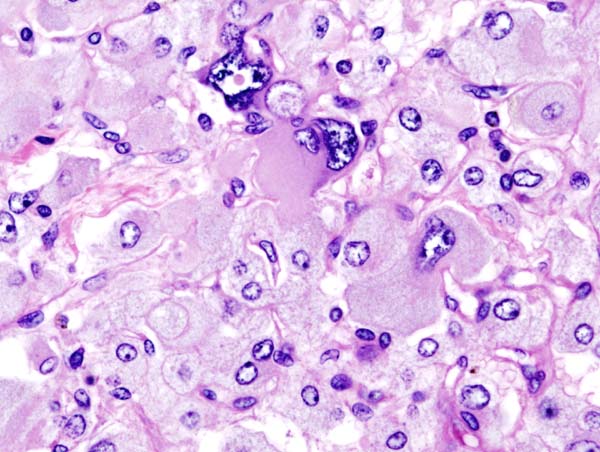
Micrograph of pheochromocytoma.
-
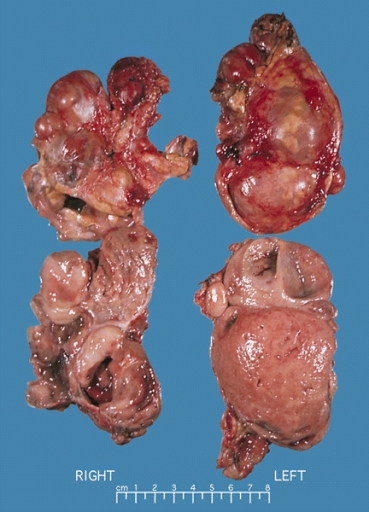
Bilateral pheochromocytoma in MEN2. Gross image.
-
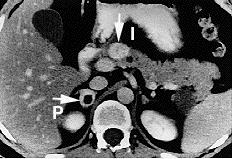
Pheochromocytoma. CT abdomen.
-
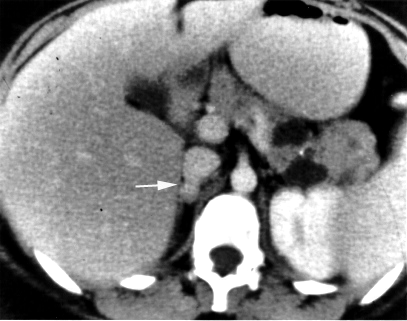
Pheochromocytoma. CT abdomen.
_histopathology.jpg)
_histopathology.jpg)



References
- ↑ Boulpaep, Emile L.; Boron, Walter F. (2003). Medical physiology: a cellular and molecular approach. Philadelphia: Saunders. p. 1065. ISBN 0-7216-3256-4.
- ↑ Online Mendelian Inheritance in Man (OMIM) MULTIPLE ENDOCRINE NEOPLASIA, TYPE IIA; MEN2A -171400
- ↑ Diouf B, Ka EH, Calender A, Giraud S, Diop TM (2000). "Association of medullary sponge kidney disease and multiple endocrine neoplasia type IIA due to RET gene mutation: is there a causal relationship?". Nephrol. Dial. Transplant. 15 (12): 2062–3. doi:10.1093/ndt/15.12.2062. PMID 11096158.
External links
- Pheochromocytoma clinical trial currently recruiting patients
- Template:MedlinePlusOverview
- overview from National Cancer Institute
- Pheochromocytoma Research Support Organization at pressor.org
- Pheochromocytoma Support Worldwide at pub1.ezboard.com
- pheochromocytoma.org
Template:Epithelial neoplasms de:Phäochromozytom it:Feocromocitoma he:פאוכרומוציטומה nl:Feochromocytoom sv:Feokromocytom