Long QT Syndrome overview: Difference between revisions
No edit summary |
|||
| Line 50: | Line 50: | ||
[[Romano-Ward syndrome]] is an [[autosomal dominant]] form of LQTS that is ''not'' associated with [[deafness]]. | [[Romano-Ward syndrome]] is an [[autosomal dominant]] form of LQTS that is ''not'' associated with [[deafness]]. | ||
==Differentiating Long QT Syndrome from Other Disorders == | |||
There are multiple causes of QT prolongation that are distinct from Long QT syndrome such as drugs (anti-arrhythmic drugs, anti-psychotic drugs), electrolyte disturbances ([[hyperkalaemia]], [[hypocalcaemia]], [[hypoglycaemia]], [[hypokalaemia]], and [[hypomagnesemia]]), neurologic events such as [[subarachnoid hemorrhage]], and [[anorexia nervosa]]. | |||
== Electrocardiography == | |||
===Electrocardiographic Examples=== | ===Electrocardiographic Examples=== | ||
<div align="left"> | <div align="left"> | ||
Revision as of 14:47, 21 October 2012
|
Long QT Syndrome Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Long QT Syndrome overview On the Web |
|
American Roentgen Ray Society Images of Long QT Syndrome overview |
|
Risk calculators and risk factors for Long QT Syndrome overview |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [2]
Overview
The long QT syndrome (LQTS) is a heart condition associated with prolongation of repolarisation (recovery) following depolarisation (excitation) of the cardiac ventricles. It is associated with syncope (fainting) and sudden death due toventricular arrhythmias. Arrhythmias in individuals with LQTS are often associated with exercise or excitement. LQTS is associated with the rare, ventricular arrhythmia torsade de pointes, which can deteriorate into ventricular fibrillation and ultimately death.
Individuals with LQTS have a prolongation of the QT interval on the ECG. The Q wave on the ECG corresponds to ventricular depolarization while the T wave corresponds to ventricular repolarization. The QT interval is measured from the Q point to the end of the T wave. While many individuals with LQTS have persistent prolongation of the QT interval, some individuals do not always show the QT prolongation; in these individuals, the QT interval may prolong with the administration of certain medications.
Classification
There are multiple genetic mutations that account for LQTs, but LQT1, LQT2, and LQT3 account for 75% of cases of LQT.
LQT1
LQT1 is the most common type of long QT syndrome, making up about 40 to 55 percent of all cases. This variant will sometimes come to the attention of the cardiologist following a cardiac event during exercise like swimming. The LQT1 gene is KCNQ1 which has been isolated to chromosome 11p15.5. KCNQ1 codes for the voltage-gated potassium channel KvLQT1 that is highly expressed in the heart. It is believed that the product of the KCNQ1 gene produces an alpha subunit that interacts with other proteins (particularly the minK beta subunit) to create the IKs ion channel, which is responsible for the delayed potassium rectifier current of the cardiac action potential.
Mutations to the KCNQ1 gene can be inherited in an autosomal dominant or an autosomal recessive pattern in the same family. In the autosomal recessive mutation of this gene,homozygous mutations in KVLQT1 leads to severe prolongation of the QT interval (due to near-complete loss of the IKs ion channel), and is associated with increased risk of ventricular arrhythmias and congenital deafness. This variant of LQT1 is known as the Jervell and Lange-Nielsen syndrome.
Most individuals with LQT1 show paradoxical prolongation of the QT interval with infusion of epinephrine. This can also unmark latent carriers of the LQT1 gene.
Many missense mutations of the LQT1 gene have been identified. These are often associated with a high risk percentage of symptomatic carriers and sudden death.
LQT2
The LQT2 type is the second most common gene location that is affected in long QT syndrome, making up about 35 to 45 percent of all cases. This variant will sometimes come to the attention of the cardiologist as a result of a cardiac event during the post partum period or after being triggered by an alarm clock. This form of long QT syndrome most likely involves mutations of the human ether-a-go-go related gene (HERG) on chromosome 7. The HERG gene (also known as KCNH2) is part of the rapid component of the potassium rectifying current (IKr). (The IKr current is mainly responsible for the termination of the cardiac action potential, and therefore the length of the QT interval.) The normally functioning HERG gene allows protection against early after depolarizations (EADs).
Most drugs that cause long QT syndrome do so by blocking the IKr current via the HERG gene. These include erythromycin, terfenadine, andketoconazole. The HERG channel is very sensitive to unintended drug binding due to two aromatic amino acids, the tyrosine at position 652 and the phenylalanine at position 656. These amino acid residues are poised so drug binding to them will block the channel from conducting current. Other potassium channels do not have these residues in these positions and are therefore not as prone to blockage.
LQT3
The LQT3 type of long QT syndrome accounts for 5-10% of cases, and cardiac events can occur during sleep. This variant involves a mutation of the gene that encodes the alpha subunit of the Na+ ion channel. This gene is located on chromosome 3p21-24, and is known as SCN5A (also hH1 and NaV1.5). The mutations involved in LQT3 slow the inactivation of the Na+ channel, resulting in prolongation of the Na+ influx during depolarization. Paradoxically, the mutant sodium channels inactivate more quickly, and may open repetitively during the action potential.
A large number of mutations have been characterized as leading to or predisposing LQT3. Calcium has been suggested as a regulator of SCN5A, and the effects of calcium on SCN5A may begin to explain the mechanism by which some these mutations cause LQT3. Furthermore mutations in SCN5A can cause Brugada syndrome, cardiac conduction disease anddilated cardiomyopathy. Rarely some affected individuals can have combinations of these diseases.
Pathophysiology
The pathophysiology of Long QT syndrome involves an inhereted or congenital abnormality in the ion channels leading to prolongation of the action potential and early after depolarization. There is also an imbalance in the sympathetic innervation of heart.
Associated syndromes
A number of syndromes are associated with LQTS.
Jervell and Lange-Nielsen syndrome
The Jervell and Lange-Nielsen syndrome (JLNS) is an autosomal recessive form of LQTS with associated congenital deafness. It is caused specifically by mutation of the KCNE1 and KCNQ1 genes
In untreated individuals with JLNS, about 50 percent die by the age of 15 years due to ventricular arrhythmias.
Romano-Ward syndrome
Romano-Ward syndrome is an autosomal dominant form of LQTS that is not associated with deafness.
Differentiating Long QT Syndrome from Other Disorders
There are multiple causes of QT prolongation that are distinct from Long QT syndrome such as drugs (anti-arrhythmic drugs, anti-psychotic drugs), electrolyte disturbances (hyperkalaemia, hypocalcaemia, hypoglycaemia, hypokalaemia, and hypomagnesemia), neurologic events such as subarachnoid hemorrhage, and anorexia nervosa.
Electrocardiography
Electrocardiographic Examples
-
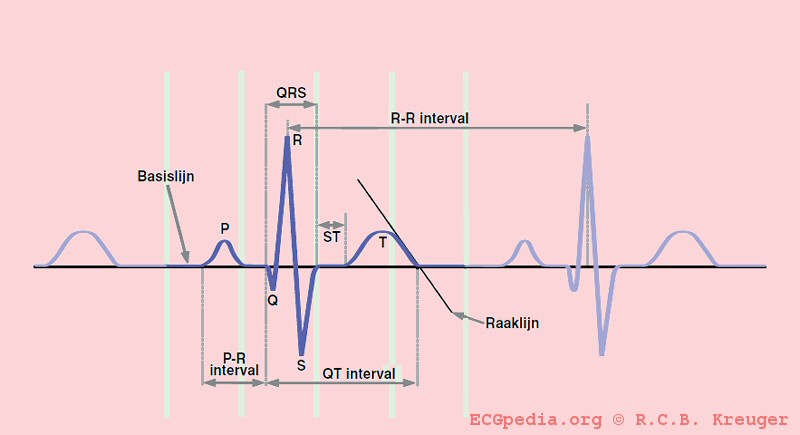
The QT interval start at the onset of the Q wave and ends where the tangent line for the steepest part of the T wave intersects with the baseline of the ECG.
-
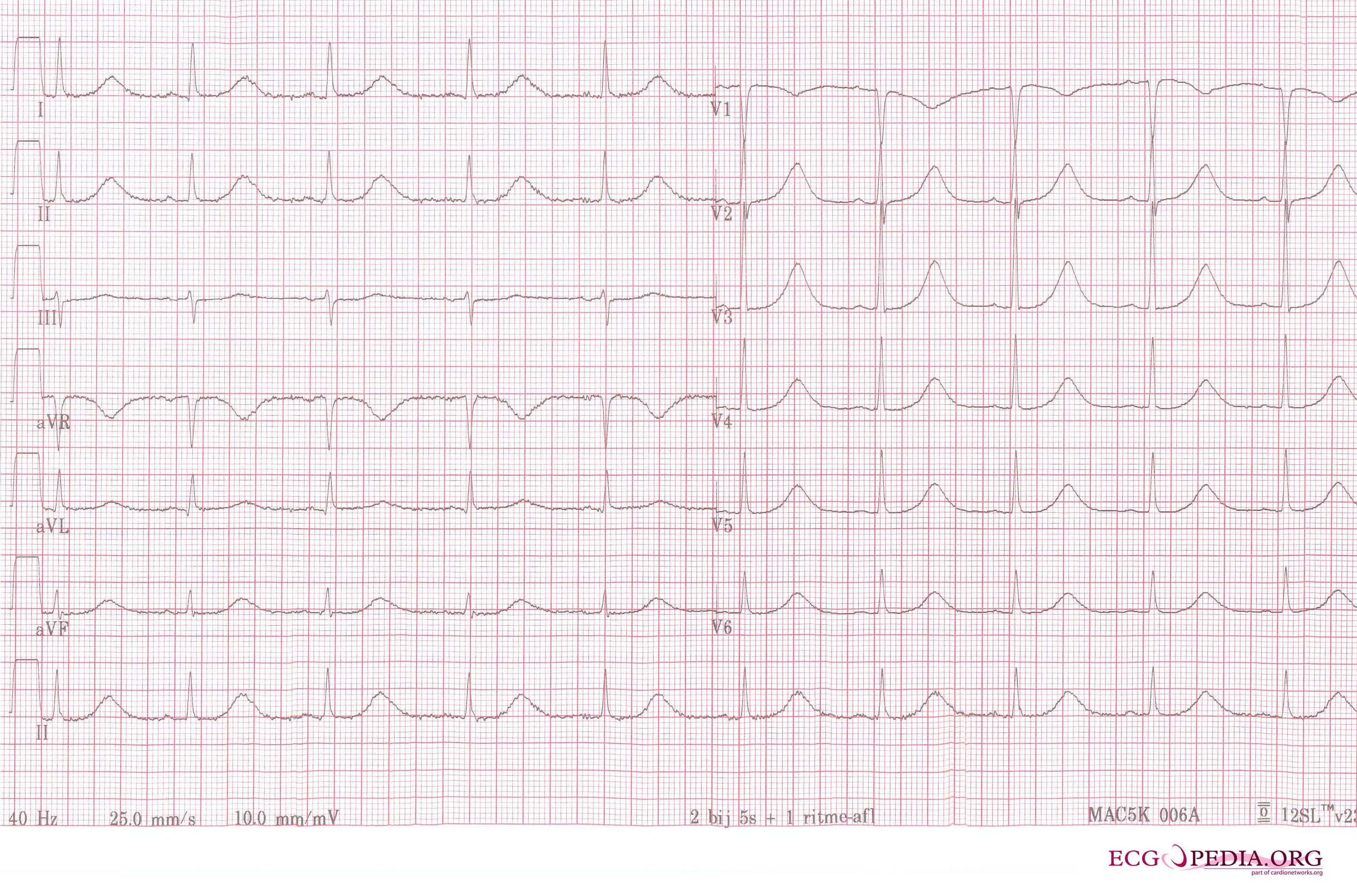
A 12 lead ECG of a patient with acquired long QT syndrome. Notice the QT prolongation. The QTc is about 640ms.
-
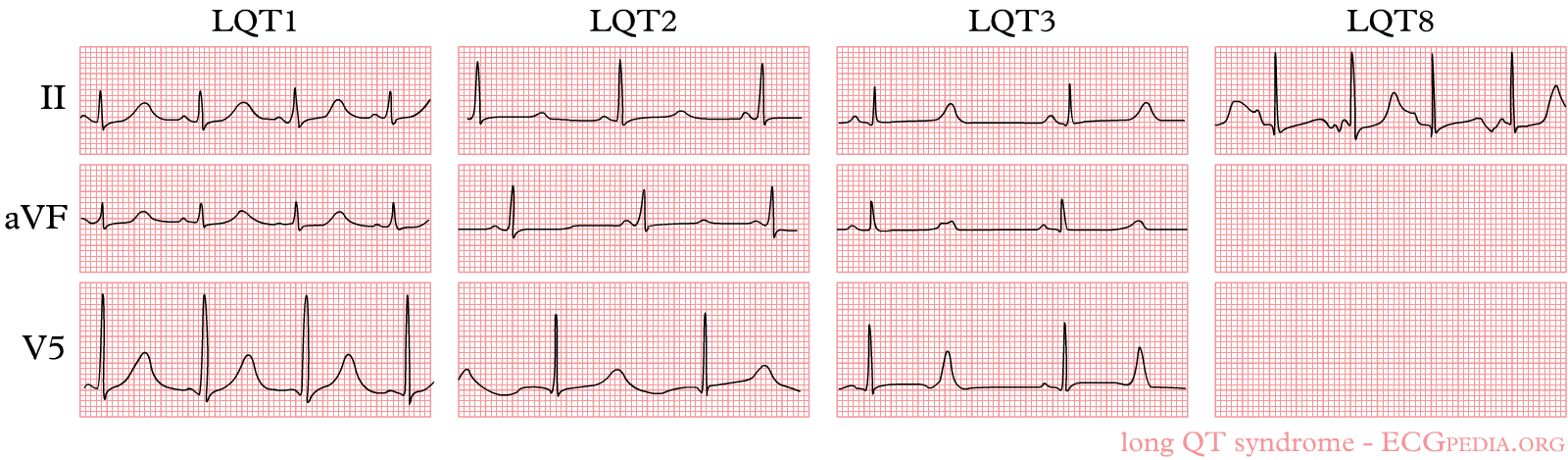
The three most common forms of LQTS can be recognized by the characteristic ECG abnormalities


-
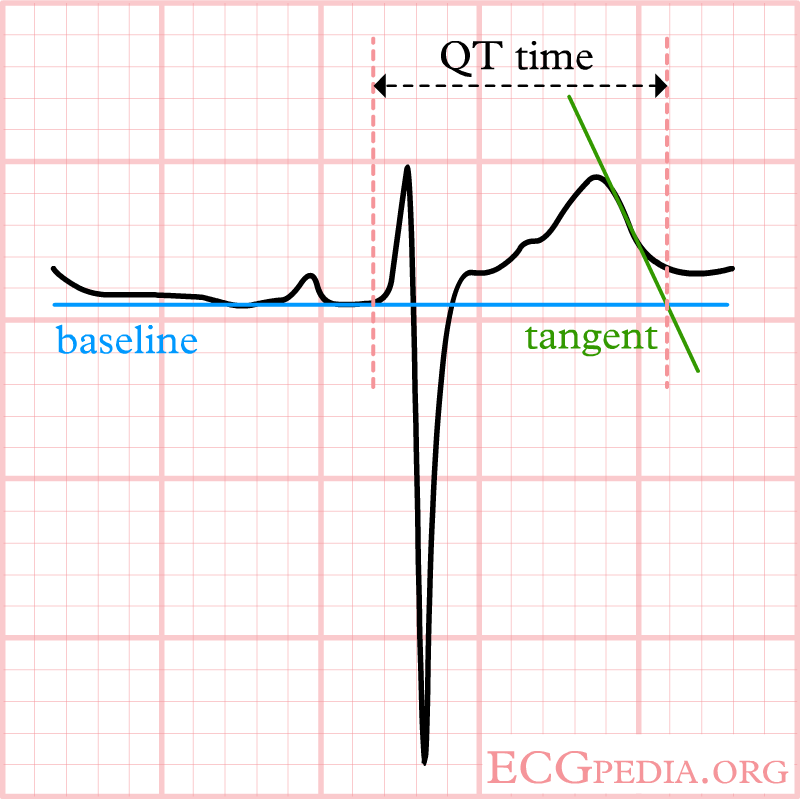
The ECG does not meet the baseline after the end of the T wave. Still, the crossing of the tangent and baseline should be used for measurements.
-
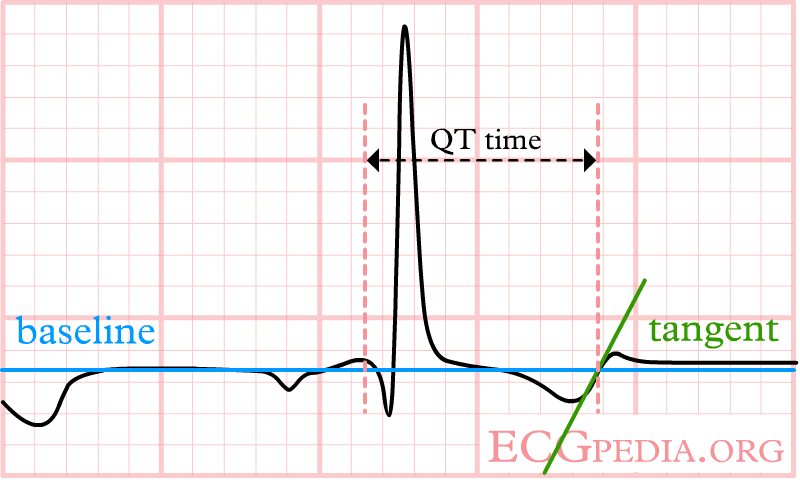
A bifasic T wave. The tangent to the 'hump' with the largest amplitude is chosen. This can change from beat to beat, making it more important to average several measurements.
-
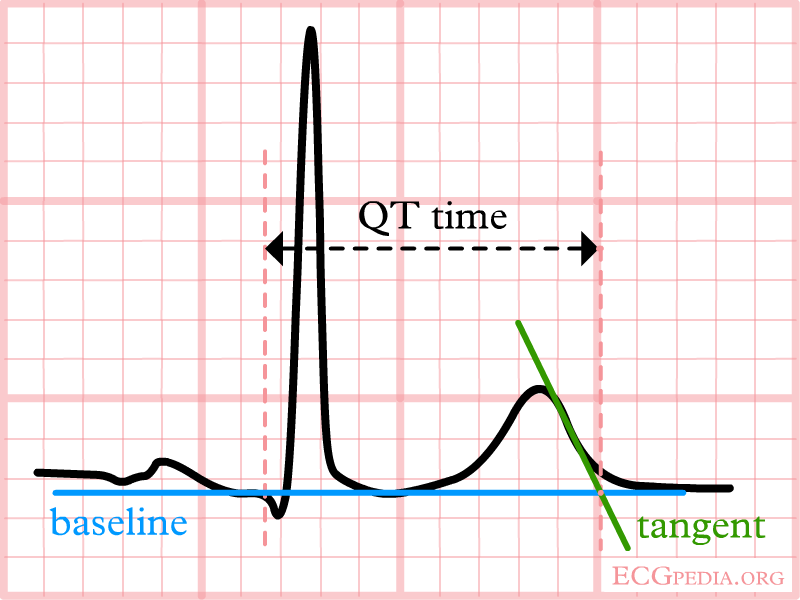
The T wave is broad, but the tangent crosses the baseline before the T wave joins the baseline. The QT interval would be overestimated when this last definition of the end of the T wave would be used.


