Chronic myelogenous leukemia pathophysiology: Difference between revisions
No edit summary |
Badria Munir (talk | contribs) |
||
| (22 intermediate revisions by 3 users not shown) | |||
| Line 1: | Line 1: | ||
__NOTOC__ | __NOTOC__ | ||
{{Chronic myelogenous leukemia}} | {{Chronic myelogenous leukemia}} | ||
{{CMG}}{{AE}} {{MJK}} | {{CMG}}{{AE}} {{Badria}} {{MJK}} {{SN}} | ||
==Overview== | ==Overview== | ||
Chronic myeloid leukemia (CML), a myeloproliferative | [[Chronic myeloid leukemia]] (CML), a [[Myeloproliferative neoplasm|myeloproliferative disorder]], which is characterized by the uncontrolled expansion of immature [[bone marrow]] [[cells]] of [[myeloid]] origin.The hallmark of [[CML]] is the formation of the [[Philadelphia chromosome]] resulting from the [[Reciprocal translocation|reciprocal translation]] (9;22)(q34;q11.2), resulting in a derivative 9q+ and a small 22q- ultimately forms a ''[[BCR/ABL]]'' [[fusion gene]] and production of a [[BCR/ABL]] [[fusion protein]]. The [[gene]] product of the ''[[BCR/ABL]]'' gene constitutively activates numerous downstream targets including ''[[c-myc]]'', ''[[Akt]]'' and ''[[Jun dimerization protein|Jun]]'', all of which cause uncontrolled proliferation and survival of [[CML]] cells. | ||
26434969/22054730 | ==Pathogenesis== | ||
* The circulating [[blood cells]] are produced in [[bone marrow]] after a series of events termed as [[hematopoiesis]].<ref name="pmid17914027">{{cite journal |vauthors=Blank U, Karlsson G, Karlsson S |title=Signaling pathways governing stem-cell fate |journal=Blood |volume=111 |issue=2 |pages=492–503 |date=January 2008 |pmid=17914027 |doi=10.1182/blood-2007-07-075168 |url=}}</ref> | |||
* The [[bone marrow]] has an tremendous [[regenerative]] ability; it is estimated that 10 trillion [[Red blood cell|red blood cells]] and 80 to 90 trillion [[leukocytes]] are formed per hour at the basal rate. | |||
* In addition to that, while cell numbers are maintained within narrow limits in normal subjects, they can be promptly increased when required. | |||
* [[Bone marrow]] primarily has small percentage of pleuripotent [[stem cells]] which give rise to various [[Progenitor cell|progenitor]] cells. | |||
* [[Hematopoiesis|Hematopoeisis]] occurs in the [[Vertebra|vertebrae]], [[pelvic bones]], [[metaphysis]] of [[long bones]] such as [[femur]], [[humerus]] in basal state. | |||
* However, during certain stressful conditions that require rapid and massive [[hematopoiesis]] such as [[thalassemia]] it then returns to its former site, [[liver]], [[spleen]] and sometimes [[lymph nodes]]. | |||
* These [[Hematopoietic stem cells|hematopoietic stem cells (HSCs]] are [[multipotent]] and have the ability to [[differentiate]] into the cells of all 10 [[blood]] lineages: | |||
** [[Erythrocytes]] | |||
** [[Platelets]] | |||
** [[Neutrophils]] | |||
** [[Eosinophils]] | |||
** [[Basophils]] | |||
** [[Monocytes]] | |||
** [[T lymphocyte|T]] and [[B lymphocytes]] | |||
** [[Natural killer cell|Natural killer cells]] | |||
** [[Dendritic cell|Dendritic cells]] | |||
* This differentiation is mediated through multiple [[growth factors]] and [[cytokines]]. <ref name="pmid16491134">{{cite journal |vauthors=Wilson A, Trumpp A |title=Bone-marrow haematopoietic-stem-cell niches |journal=Nat. Rev. Immunol. |volume=6 |issue=2 |pages=93–106 |date=February 2006 |pmid=16491134 |doi=10.1038/nri1779 |url=}}</ref> <ref name="pmid179140272">{{cite journal |vauthors=Blank U, Karlsson G, Karlsson S |title=Signaling pathways governing stem-cell fate |journal=Blood |volume=111 |issue=2 |pages=492–503 |date=January 2008 |pmid=17914027 |doi=10.1182/blood-2007-07-075168 |url=}}</ref> | |||
* The [[Hematopoietic stem cells|hematopoietic stem cells (HSCs]]) and [[progenitor cells]] are supported by a [[stromal]] cell network that provides cell-cell contact support. | |||
* The [[stromal]] network provides two major functions: | |||
* An [[Adhesion molecule|adhesive framework]] onto which the developing [[cells]] are bound, these cells produce: | |||
** A variety of [[Cell adhesion molecule|adhesion molecules]]. | |||
** [[Hematopoietic]] [[Growth factors]] or [[Cytokine|cytokines]] that are thought to support the [[Survival function|survival]], [[proliferation]], and [[differentiation]] of [[Hematopoietic stem cells|HSCs]] and [[progenitors]]. <ref name="pmid12598852">{{cite journal |vauthors=Smith C |title=Hematopoietic stem cells and hematopoiesis |journal=Cancer Control |volume=10 |issue=1 |pages=9–16 |date=2003 |pmid=12598852 |doi=10.1177/107327480301000103 |url=}}</ref> | |||
* [[Primitive (integral)|Primitive]] [[Mesenchymal stem cell|mesenchymal stromal cells]] ([[Mesenchymal stem cell|MSCs]]) are thought to have the capacity to differentiate into following: | |||
** Osteolineage cells | |||
** [[Chondrocytes]] | |||
** [[Adipocytes|Adipocyte]]<nowiki/>s | |||
** [[Perivascular cell|Perivascular cells]] | |||
* Overall Differentiation of [[myeloid]] [[progenitors]] is mediated through:<ref name="pmid25814077">{{cite journal |vauthors=Chereda B, Melo JV |title=Natural course and biology of CML |journal=Ann. Hematol. |volume=94 Suppl 2 |issue= |pages=S107–21 |date=April 2015 |pmid=25814077 |doi=10.1007/s00277-015-2325-z |url=}}</ref> | |||
** The production of essential [[hematopoietic]] [[growth factors]]. | |||
** Several [[Signaling pathway|signaling pathways]] have come up as integral control devices of [[Hematopoietic stem cell transplantation|HSC]] fate, such as:<ref name="pmid179140273">{{cite journal |vauthors=Blank U, Karlsson G, Karlsson S |title=Signaling pathways governing stem-cell fate |journal=Blood |volume=111 |issue=2 |pages=492–503 |date=January 2008 |pmid=17914027 |doi=10.1182/blood-2007-07-075168 |url=}}</ref> | |||
*** [[Notch]] | |||
*** Wingless-type [[Wnt signaling pathway|(Wnt]]) | |||
*** [[Sonic hedgehog]] ([[Sonic hedgehog|Shh]]) | |||
*** [[Smad|Smad pathways]] | |||
* These signaling circuits provide an important structure for our understanding of HSC regulation, alongwith providing information of how the [[bone marrow]] micro environment couples and integrates extrinsic with intrinsic factors responsible for [[Hematopoiesis lineages|HSC differentiation]] and development of [[chronic myeloid leukemia]].<ref name="pmid125988522">{{cite journal |vauthors=Smith C |title=Hematopoietic stem cells and hematopoiesis |journal=Cancer Control |volume=10 |issue=1 |pages=9–16 |date=2003 |pmid=12598852 |doi=10.1177/107327480301000103 |url=}}</ref> | |||
=====Genetic Translocation:===== | |||
* [[Chronic myeloid leukemia]] ([[CML]]), a [[myeloproliferative neoplasm]], characterized by the presence of the [[Philadelphia chromosome]] which is thought to be a definitive diagnostic marker for [[CML]].<ref name="pmid26434969">{{cite journal |vauthors=Thompson PA, Kantarjian HM, Cortes JE |title=Diagnosis and Treatment of Chronic Myeloid Leukemia in 2015 |journal=Mayo Clin. Proc. |volume=90 |issue=10 |pages=1440–54 |date=October 2015 |pmid=26434969 |pmc=5656269 |doi=10.1016/j.mayocp.2015.08.010 |url=}}</ref> | |||
* In [[Philadelphia chromosome]] translocation, parts of two chromosomes (the 9<sup>th</sup> and 22<sup>nd</sup> by conventional [[karyotype|karyotypic]] numbering) switch places. | |||
* As a result, part of the ''[[BCR]]'' ("breakpoint cluster region") [[gene]] from [[chromosome 22]] is fused with the ''[[Abl gene|ABL]] (''"[[Abelson murine leukemia virus|abelson murine leukemia]]"'')'' [[gene]] on [[chromosome 9]]. | |||
* This abnormal "[[Fusion gene|fusion]]" [[gene]] generates a protein of p210 . | |||
* Because ''[[ABL]]'' carries a domain that can add [[phosphate]] groups to [[tyrosine]] residues (a [[tyrosine kinase]]), the ''[[BCR/ABL]]'' [[fusion gene]] product is also a [[tyrosine kinase]]. | |||
* The fused ''[[BCR/ABL]]'' protein interacts with the [[interleukin 3]] beta c receptor subunit. | |||
* The ''[[BCR/ABL]]'' transcript is continuously active and does not require activation by other cellular messaging proteins that promotes [[growth]] and [[replication]] through downstream pathways such as: | |||
** [[RAS]] | |||
** RAF | |||
** [[JunD|JUN kinase]] | |||
** [[Myc|MYC]] | |||
** [[STAT protein|STAT]] | |||
* In turn [[BCR/ABL]] triggers a cascade of proteins which control the [[cell cycle]], speeding up [[cell division]]. | |||
* Moreover the [[BCR/ABL]] protein inhibits [[DNA repair]], causing [[genomic instability]] and making the cell more susceptible to developing further genetic [[mutations]]. | |||
* The action of the [[BCR/ABL]] protein is the pathophysiologic cause of [[chronic myelogenous leukemia]].<ref name="pmid22054730">{{cite journal |vauthors=Jabbour E, Parikh SA, Kantarjian H, Cortes J |title=Chronic myeloid leukemia: mechanisms of resistance and treatment |journal=Hematol. Oncol. Clin. North Am. |volume=25 |issue=5 |pages=981–95, v |date=October 2011 |pmid=22054730 |pmc=4428141 |doi=10.1016/j.hoc.2011.09.004 |url=}}</ref><ref name="Hehlmann">{{cite journal|title=Chronic myeloid leukaemia|author=Hehlmann R, Hochhaus A, Baccarani M; European LeukemiaNet|journal=Lancet|volume=370|issue=9584|pages=342-50|date=2007|pmid=17662883}}</ref><ref name="pmid24729196">{{cite journal |vauthors=Jabbour E, Kantarjian H |title=Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management |journal=Am. J. Hematol. |volume=89 |issue=5 |pages=547–56 |date=May 2014 |pmid=24729196 |doi=10.1002/ajh.23691 |url=}}</ref><ref name="pmid26625737">{{cite journal |vauthors=Kaleem B, Shahab S, Ahmed N, Shamsi TS |title=Chronic Myeloid Leukemia--Prognostic Value of Mutations |journal=Asian Pac. J. Cancer Prev. |volume=16 |issue=17 |pages=7415–23 |date=2015 |pmid=26625737 |doi= |url=}}</ref><ref name="pmid26434969">{{cite journal |vauthors=Thompson PA, Kantarjian HM, Cortes JE |title=Diagnosis and Treatment of Chronic Myeloid Leukemia in 2015 |journal=Mayo Clin. Proc. |volume=90 |issue=10 |pages=1440–54 |date=October 2015 |pmid=26434969 |pmc=5656269 |doi=10.1016/j.mayocp.2015.08.010 |url=}}</ref><ref name="pmid22054730">{{cite journal |vauthors=Jabbour E, Parikh SA, Kantarjian H, Cortes J |title=Chronic myeloid leukemia: mechanisms of resistance and treatment |journal=Hematol. Oncol. Clin. North Am. |volume=25 |issue=5 |pages=981–95, v |date=October 2011 |pmid=22054730 |pmc=4428141 |doi=10.1016/j.hoc.2011.09.004 |url=}}</ref> | |||
==== Role of reactive oxygen species: ==== | |||
* Recent studies have demonstarated that [[BCR/ABL]] also stimulates the production of [[reactive oxygen species]] ([[Reactive oxygen species|ROS]]), which levels increase with [[CML]] progression and this in turn increases [[BCR/ABL]] self-mutagenesis. | |||
* [[Tyrosine kinase inhibitor]] resistance can also be related to higher [[Reactive oxygen species|ROS]] production. | |||
* Therefore, [[ROS1|ROS]]-induced self-mutagenesis of [[BCR/ABL]] is of prime significance for [[CML]] progression. | |||
* These can be dependent on [[DNA repair]], which is modulated by [[BCR/ABL]] and can be different in [[CML]] stem and [[Progenitor cell|progenitor cells]].<ref name="pmid27904889">{{cite journal |vauthors=Antoszewska-Smith J, Pawlowska E, Blasiak J |title=Reactive oxygen species in BCR-ABL1-expressing cells - relevance to chronic myeloid leukemia |journal=Acta Biochim. Pol. |volume=64 |issue=1 |pages=1–10 |date=2017 |pmid=27904889 |doi=10.18388/abp.2016_1396 |url=}}</ref> | |||
===== Altered bone marrow pathway signalling: ===== | |||
* Implication of altered [[bone marrow]] signalling on [[stem cell]] persistence in the [[bone marrow]] [[Niche cell|niche]]. | |||
* Recent advancements have been trying to establish the relationship between [[bone marrow]] pathway and [[Wnt signaling pathway|Wnt pathways]] and their role to alter the Cdx-Hox axi.<ref name="pmid27911727">{{cite journal |vauthors=Toofan P, Wheadon H |title=Role of the bone morphogenic protein pathway in developmental haemopoiesis and leukaemogenesis |journal=Biochem. Soc. Trans. |volume=44 |issue=5 |pages=1455–1463 |date=October 2016 |pmid=27911727 |doi=10.1042/BST20160104 |url=}}</ref> | |||
====Role of Integrin:==== | |||
* CML cells present in contact with stroma or fibronectin continue to [[proliferate]], suggesting that failure to adhere through [[integrin]] receptors may also underlie the abnormal proliferation of [[CML]] progenitors.<ref name="VerfaillieHurley1997">{{cite journal|last1=Verfaillie|first1=Catherine M.|last2=Hurley|first2=Randolph|last3=Zhao|first3=Robert C.H.|last4=Prosper|first4=Felipe|last5=Delforge|first5=Michel|last6=Bhatia|first6=Ravi|title=Pathophysiology of CML: Do defects in integrin function contribute to the premature circulation and massive expansion of the BCR/ABL positive clone?|journal=Journal of Laboratory and Clinical Medicine|volume=129|issue=6|year=1997|pages=584–591|issn=00222143|doi=10.1016/S0022-2143(97)90192-X}}</ref> | |||
* Although, [[CML]] [[progenitors]] express the same [[Integrin|integrin receptors]] as normal [[progenitors]], they fail to adhere to [[stroma]] and [[fibronectin]]. | |||
* This indicates that structural or functional abnormalities of these [[receptors]] can be integral part of pathogenesis. | |||
== | ====Blast crisis:==== | ||
=== | * [[Chronic myelogenous leukemia|Chronic myeloid leukemia (CML)]] in [[blast crisis]] is the transition of [[CML]] in chronic or accelerated phase to an [[acute leukemia]]. | ||
Chronic myeloid leukemia (CML) | * It is characterized by: | ||
** ≥ 30% [[blast]]s in the [[bone marrow]] or peripheral [[blood]]. | |||
** The development of extramedullary disease outside of the [[spleen]]. | |||
* In light of recent changes in the World Health Organization, definition of acute leukemia, the percentage of [[blast]]s required for [[CML]] in blastic phase may someday be reduced to 20%.<ref name="pmid6968038">{{cite journal |vauthors=Martin PJ, Najfeld V, Hansen JA, Penfold GK, Jacobson RJ, Fialkow PJ |title=Involvement of the B-lymphoid system in chronic myelogenous leukaemia |journal=Nature |volume=287 |issue=5777 |pages=49–50 |date=September 1980 |pmid=6968038 |doi= |url=}}</ref> | |||
* Consistent with the early [[stem cell]] nature of [[CML]], blastic transformation may be: | |||
** [[Myeloid]] | |||
** [[Lymphoid]] | |||
** Undifferentiated/mixed | |||
* Myeloid blast crisis being about two times more common than lymphoid. | |||
* It is suggested that [[blast crisis]] is due to one of following reasons:<ref name="pmid10942235">{{cite journal |vauthors=Salloukh HF, Laneuville P |title=Increase in mutant frequencies in mice expressing the BCR-ABL activated tyrosine kinase |journal=Leukemia |volume=14 |issue=8 |pages=1401–4 |date=August 2000 |pmid=10942235 |doi= |url=}}</ref> | |||
** [[BCR/ABL]] is considered to be responsible for progressive [[genomic instability]] or [[epigenetic changes]], which occur at the [[CML]] stem cell level and/or in later [[CML]] [[progenitor cells]]. | |||
** The degree of [[genomic instability]] is directly related to the level of [[BCR/ABL]] [[kinase]] activity. | |||
** The third is that [[CML]] [[stem cells]] are the least vulnerable to [[ABL]]-targeted therapy and may serve as reservoirs for [[CML]] progression. | |||
* All these events concomitantly result in the acquired loss of [[hematopoietic cell]] [[differentiation]], resulting in a highly progressive generation of immature [[Blast|blasts]] in peripheral [[blood]] and in [[bone marrow]]. | |||
* Various studies have [[BCR/ABL]] is implicated in the generation and maintenance of secondary [[DNA]] alterations. | |||
====== Genetic Alterations in Blast crisis: ====== | |||
* Following [[genetic]] changes have been observed which play crucial role in progression of disease phase. | |||
** Duplication of the [[Philadelphia chromosome|Ph chromosome]], [[trisomy 8]], and [[isochromosome]] 17.<ref name="pmid3477958">{{cite journal |vauthors=Kantarjian HM, Keating MJ, Talpaz M, Walters RS, Smith TL, Cork A, McCredie KB, Freireich EJ |title=Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients |journal=Am. J. Med. |volume=83 |issue=3 |pages=445–54 |date=September 1987 |pmid=3477958 |doi= |url=}}</ref> | |||
** Alterations in [[P53 (protein)|p53]] have been found in only a minority of cases.<ref name="AhujaBar-Eli1989">{{cite journal|last1=Ahuja|first1=H.|last2=Bar-Eli|first2=M.|last3=Advani|first3=S. H.|last4=Benchimol|first4=S.|last5=Cline|first5=M. J.|title=Alterations in the p53 gene and the clonal evolution of the blast crisis of chronic myelocytic leukemia.|journal=Proceedings of the National Academy of Sciences|volume=86|issue=17|year=1989|pages=6783–6787|issn=0027-8424|doi=10.1073/pnas.86.17.6783}}</ref> | |||
** Loss of p16INK4A/ARF has been reported in up to half of patients with CML in [[lymphoid]] [[blast crisis]] but is rare in the [[myeloid]] form.<ref name="pmid12563607">{{cite journal |vauthors=Hasford J, Pfirrmann M, Hehlmann R, Baccarani M, Guilhot F, Mahon FX, Kluin-Nelemans HC, Ohnishi K, Thaler J, Steegmann JL |title=Prognosis and prognostic factors for patients with chronic myeloid leukemia: nontransplant therapy |journal=Semin. Hematol. |volume=40 |issue=1 |pages=4–12 |date=January 2003 |pmid=12563607 |doi=10.1053/shem.2003.50006 |url=}}</ref> | |||
** Thus, it is hypothesized that clonal evolution plays integral role in blastic progression and is likely facilitated by the dysregulation of normal [[apoptotic]] pathways by [[BCR/ABL]]. | |||
* Recent studies have implicated activation of the following pathways <ref name="Donato2003">{{cite journal|last1=Donato|first1=N. J.|title=BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571|journal=Blood|volume=101|issue=2|year=2003|pages=690–698|issn=00064971|doi=10.1182/blood.V101.2.690}}</ref> | |||
** [[LYN]] | |||
** [[AKT]] | |||
** [[STAT5]] | |||
=== Gross Pathology === | |||
On gross pathology, no distinctive findings are seen in [[chronic myeloid leukemia]]. | |||
=== Microscopic Pathology === | |||
[[Blast]] cells are seen on peripheral blood smear of patients of [[chronic myeloid leukemia]] which are present during [[blast crisis]]. | |||
<gallery widths="200px" class="center"> | |||
CML.jpg| Blast crisis of chronic myelogenous leukemia (CML). Peripheral blood smear revealing the histopathologic features indicative of a blast crisis in the case of chronic myelogenous leukemia.<ref name="cdc">Center for Disease Control and Prevention. Public Health Image Library 2015.http://phil.cdc.gov/phil/details_linked.asp?pid=6</ref> | |||
</gallery> | |||
==References== | ==References== | ||
Latest revision as of 05:30, 31 January 2019
|
Chronic myelogenous leukemia Microchapters |
|
Differentiating Chronic myelogenous leukemia from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Chronic myelogenous leukemia pathophysiology On the Web |
|
American Roentgen Ray Society Images of Chronic myelogenous leukemia pathophysiology |
|
Directions to Hospitals Treating Chronic myelogenous leukemia |
|
Risk calculators and risk factors for Chronic myelogenous leukemia pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]Associate Editor(s)-in-Chief: Badria Munir M.B.B.S.[2] Mohamad Alkateb, MBBCh [3] "sandbox:SN"
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [4]; Associate Editor(s)-in-Chief:
Overview
Pernicious anemia (also called Addison's anemia) is a type of red blood cell disorder caused by impaired vitamin B12 metabolism. Vitamin B12 is primarily absorbed by the small intestine, after being bound to intrinsic factor secreted by parietal cells of gastric mucosa. When this process is disrupted by conditions like atrophic gastritis, celiac disease, small bowel resection etc, B12 deficiency ensues.
Historical perspective
- Pernicious anemia was first discovered by Thomas Addison, hence it is also known as addison's anemia.
- Loss of life from large volume blood loss in the people fighting in the first world war inspired George Whipple to investigate blood forming components such as arsenic, iron pills etc, but found liver to be the most effective. He bled dogs until they had clinical anemia and fed them cooked liver which showed an improvement in symptoms and hematopoeisis. [1]
- In 1948, Smith, Rickles et al., isolated the anti-pernicious factor from liver extract and named it Vitamin B12. They showed that even small amounts of this factor can be used to treat and to prevent pernicious anemia. [2]
Pathophysiology
Vitamin B12 is an essential vitamin for humans and animals because we cannot synthesise it on our own. B12 is a cofactor in DNA synthesis and other important biochemical reactions. Vitamin B12 deficiency manifests as anemia because hematopoetic stem cells in the bone marrow which are rapidly dividing need B12 for division and DNA production. This process is impaired leading to ineffective hematopoeisis. Vitamin B12 is also necessary for production of myelin which is an important component in the covering sheath of nerves. Deficiency results in improper nerve conduction due to nerve destabilisation. [3]
Physiology
- Vitamin B12 is also called cobalamin because it contains cobalt at the core of its structure. Dietary sources of vitamin B12 include meat, fish and eggs.[4]
- When consumed through its dietary source, B12 is bound to protein till it enters the stomach.
- In the stomach, B12 is uncoupled from its carrier protein due to the presence of gastric acid, which is why vitamin B12 deficiency is so commonly seen among those on chronic antacid medication. [5]
- Once in the stomach, it is then bound to gastric R binder, a glycoprotein secreted by the salivary glands till it reaches the duodenum.[6]
- In the duodenum and jejunum, the pancreatic enzymes digest the gastric R binder and cobalamin is bound to intrinsic factor (IF).
- Intrinsic factor is secreted by the gastric parietal cells. Once bound to IF, vitamin B12 travels up to the ileum where IF is removed and B12 binds with carrier proteins called transcobalamins and this complex is taken up by the liver and bone marrow, among other tissues.
- Inside the cells, the transcobalamin-B12 complex is dissolved and cobalamin is reduced to methylcobalamin which serves as a cofactor and coenzyme in many important biochemical reactions[7].
The two major reactions involving B12 in the human body are:
- Vitamin B12 in the from of cyanocobalamin is required in the synthesis of methionine. Methionine is produced from homocysteine and is catalysed by the enzyme methionine synthase. This enzyme utilises cyanocobalamin as a cofactor. Deficiency of vitamin B12 causes a decreased production of methionine and buildup of homocysteine. Hyperhomocysteinemia is implicated as a risk factor in cardiovascular disease.[8]
- The Kreb's cycle utilises vitamin B12 in the reaction converting methylmalonyl-CoA to succinyl-CoA. Thus vitamin B12 deficiency causes a buildup of methylmalonic acid, the substrate for the enzyme methylmalonyl coenzyme A mutase. Methylmalonic acid levels are elevated in the urine of people affected with pernicious anemia and other forms of B12 deficiency.
Storage
The human body can store anywhere from 2-5mg of vitamin B12. Most of this is stored in the liver and is recycled via enterohepatic circulation.
Pathogenesis
Pernicious anemia is a type of megaloblastic anemia caused due to improper vitamin B12 absorption by the body. Impaired absorption occurs because of deficiency of intrinsic factor which is produced by the parietal cells of the stomach. The etiology of pernicious anemia can be due to autoimmune causes or genetic disease. In autoimmune disease, the antibodies attack most of the gastric mucosa, but the antrum is spared.
Autoimmune causes of pernicious anemia
This is the most common cause of pernicious anemia. In autoimmune pernicious anemia, the body produces antibodies against parietal cells or intrinsic factor.
- Antibodies against parietal cells of the gastric mucosa work to inhibit the H+/K(+)-ATPase which is the proton pump present in the parietal cells. The proton pump serves as an auto antigen and activates the cytotoxic CD4+ T cells which proceed to destroy gastric mucosal cells.[9][10]
- Intrinsic factor antibodies are present in fewer cases of pernicious anaemia but are highly specific. There are 2 types of IF antibodies. They prevent the binding and absorption of cobalamin in the ileum via its receptor.[11]
Clinical features
- The symptoms of pernicious anemia take months, and often years to manifest. Patients most commonly present with symptoms of anemia like lightheadedness, dizziness, shortness of breath etc. The population affected with pernicious anemia is usually the elderly (>60 years) owing to its insidious onset.
- Pernicious anemia has hematological, gastrointestinal and neurological manifestations.
- Hematological signs are the earliest manifestation of the disease while neurological signs are seen much later.
- Patients with pernicious anemia usually have very low levels of hydrochloric acid in the stomach (achlorhydria) and high levels on gastrin (hypergastrinemia).
Differentiating pernicious anemia from other diseases
Pernicious anemia shares many similarities with other forms of megaloblastic anemia like B12 and folate deficiency.
- Vitamin B12 deficiency due to insufficient intake (eg veganism) has all the features of pernicious anemia like megaloblasts, hypersegmented neutrophils, neuropsychiatric manifestations. But atrophic gastritis is absent, so achlorhydria, parietal cell antibodies or IF antibodies are absent. Intrinsic factor levels are also normal.[6]
- Folic acid deficiency also results in megaloblastic anemia and similar hematological changes as pernicious anemia, but urinary excretion of methylmalonic acid is absent, so are features of pernicious anemia like achlorhydria, antibodies and normal IF levels.
- Ileal resection causes B12 deficiency due to decreased absorption.
- Certain drugs such as methotrexate, azathioprine cause folate deficiency and result in megaloblastic anemia. This is usually seen in patients taking chemotherapy or other chronic conditions such as rheumatoid arthritis. [12]
- Chronic proton pump inhibitor therapy also results in B12 deficiency as vitamin B12 cannot dissociate from its carrier protein in the absence of an acidic environment.[13]
- Long term use of metformin, such as in diabetics, is linked to vitamin B12 deficiency and symptoms similar to pernicious anemia, but this can be differentiated from pernicious anemia as it is seen in diabetics on chronic therapy.[14]
Associated Conditions
People affected with pernicious anemia might have other coexisting autoimmune conditions such as autoimmune thyroiditis, autoimmune diabetes, vitiligo etc. Autoimmune thyroiditis is most commonly seen in patients with pernicious anemia, particularly females. HLA DR3 has been implicated in the development of autoimmune diseases such as pernicious anemia[15].
Epidemiology and demographics
- Pernicious anemia is a disease of the elderly. The mean age of patients who are symptomatic is >60.[16]
- An exception is the genetic form of the disease which is a congenital deficiency of intrinsic factor and is seen in children <10 years of age.
- Men and women are equally affected
- Prevalence of pernicious anemia is estimated at 0.1% of the population.[17]
Genetics
- Some forms of pernicious anemia are congenital and a genetic link has been postulated because of a higher incidence in certain populations.
- Affected people have a complete or near total absence of intrinsic factor and the presence of antibodies against intrinsic factor.
- The genetic variant is transmitted through an autosomal recessive pattern.[18]
Risk factors
- People who have autoimmune conditions like diabetes mellitus, autoimmune thyroiditis are at higher risk of developing pernicious anemia.
Natural History, Complications and Prognosis
- In most cases, patients affected with pernicious anemia remain asymptomatic for many years.
- Early manifestations include fatigue, shortness of breath, pallor and weakness.
- Long standing untreated pernicious anemia results in irreversible neurological damage such as subacute combined degeneration of the spinal cord.
- Neurological changes are irreversible once they set in and do not resolve with cobalamin supplementation.
Diagnosis
A diagnosis of pernicious anemia is made by a history and physical examination, along with hematological and neurological examination.
Diagnostic criteria
- The only specific criteria to diagnose pernicious anemia is an intrinsic factor output of less than 200U/h after pentagastrin stimulation, where normal levels would be >2000U/h. [19]
Symptoms
Symptoms of pernicious anemia are summarised below
| Hematological symptoms | Gastrointestinal symptoms | Neurological symptoms |
|---|---|---|
| Fatigue | Loss of appetite | Parasthesias |
| Weakness | Weight loss
|
Depression |
| Shortness of breath | Nausea | Gait problems |
| Dizziness | Burning sensation on tongue | Weakness |
| Tachycardia | Diarrhea | Loss of balance |
| Lightheadedness | Vomiting | Confusion |
Physical examination findings
Most important physical examination findings are the neurological findings of long standing B12 deficiency which leads to subacute combined degeneration of the spinal cord.
- Hematological signs include pallor and icterus.[20]
- Neurological signs: Vitamin B12 deficiency causes nerve demyelination. B12 deficiency also causes a buildup of methylmalonic acid which is toxic to neuronal cells and causes apoptosis.[21].
The main neurological manifestation of pernicious anemia and vitamin B12 deficiency is subacute combined degeneration. The posterior and lateral columns of the spinal cord are affected. Lateral column demyelination manifests as hyperreflexia and spasticity, while posterior column defects are loss of proprioception and vibration sense. Ataxia and loss of tandem gait are also manifestations of posterior column demyelination. Recreational or accidental inhalation of nitrous oxide gas (laughing gas) can precipitate subacute combined degeneration in people with low levels of vitamin B12.[22]
- Gastrointestinal signs: Upto 25% of people affected with pernicious anemia develop glossitis. The tongue appears red, "beefy" and smooth due to atrophy and blunting of the lingual papillae.[23]
Subacute combined degeneration

Laboratory findings
- The first step in diagnosis is a blood vitamin B12 level. Blood levels less than 200 pg/ml are seen in pernicious anemia.
- Intrinsic factor antibodies and Parietal cell antibodies.
- Low intrinsic factor level.[24]
- Gastric mucosal sampling shows parietal cell atrophy with antral sparing.[25]
- Increased level of gastrin.
- Increased levels of homocysteine and methylmalonyl-CoA.
- Decreased folate levels are seen due to "folate trapping" in the form of methyltetrahydrofolate.
Shilling Test
The Shilling test is no longer done to detect an IF deficiency but has historical importance. After a vitamin B12 deficiency is noted, the patient is given radioactively tagged cobalamin to take orally. Soon after this step, the patient is injected with unlabelled cobalamin intramuscularly. Urine is checked for radioactive cobalamin for the next 24 hours. In pernicious anemia, there is an intrinsic factor deficiency, therefore the orally consumed radioactive cobalamin will not be absorbed and can be detected in the urine. In the next step, the patient is given radioactive cobalamin along with intrinsic factor and their urine is checked for traces of radioactive cobalamin. Absence of radioactive cobalamin in the urine points to the deficiency of intrinsic factor in the patients stomach which is the cause of vitamin B12 deficiency[26]. If the cobalamin absorption does not increase even with intrinsic factor supplementation, patient can be given a course of antibiotics as bacterial overgrowth may hinder absorption.
Peripheral smear findings
- The most obvious peripheral smear finding is megaloblasts and macrocytes.
Megaloblastic anemia results due to the lagging behind of nuclear development when compared to cytoplasmic development. This is known as nuclear-cytoplasmic asynchrony. Such defective cells are destroyed in the bone marrow (intramedullary hemolysis).
- Decreased number of RBCs (erythopenia)
- Macrocytosis- the RBCs in pernicious anemia are very large. Macrocytosis is defined as cells that have an MCV >100 femtolitres (normal :80-100fL)
- Hypersegmented neutrophils : Neutrophils containing ≥ 6 lobes. [27]
- Poikilocytosis and anisocytosis
- Low reticulocyte count (reticulopenia)
- Howell-Jolly bodies
-
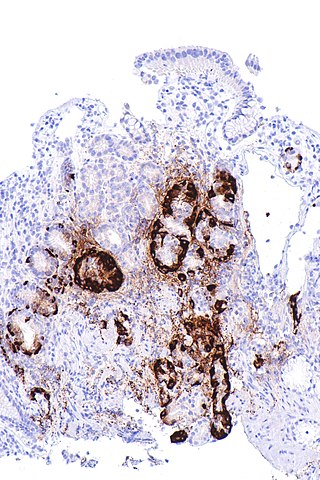
Atrophic gastritis
-
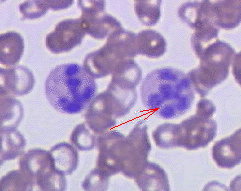
Hypersegmented neutrophil


Treatment
- Standard treatment for pernicious anemia is replacement of cobalamin via intramuscular injection. [28]
- 1000 mcg IM everyday for one week, followed by weekly injections the next month and then monthly once injections.
- Response to treatment is measured by an increase in reticulocyte count within 5 days of starting therapy.
- Patient also experience a sense of wellbeing shortly after beginning therapy.
- If reticulocytosis is not observed within the first week of therapy, other factors such as hypothyroidism, folate deficiency should be considered.
- Intramuscular therapy can be replaced by high dose oral therapy.[17]
- Neurological disease always warrants parenteral treatment.
- Within the first 3-4 weeks of treatment, marrow changes revert and there is resolution in macrocytosis.
- Most patients require lifelong monthly therapy.
- Routine follow up should be done with a CBC every few months.
- A small percentage of patients develop gastric carcinoma, particularly in the elderly. Regular surveillance helps in early detection and treatment. [29]
Prevention
- There is no primary preventive measure for pernicious anemia.
- Once sucessfully diagnosed and treated, patients with pernicious anemia are followed up every year for development of stomach cancer[30], or symptoms of anemia.
References
Overview
Chronic myeloid leukemia (CML), a myeloproliferative disorder, which is characterized by the uncontrolled expansion of immature bone marrow cells of myeloid origin.The hallmark of CML is the formation of the Philadelphia chromosome resulting from the reciprocal translation (9;22)(q34;q11.2), resulting in a derivative 9q+ and a small 22q- ultimately forms a BCR/ABL fusion gene and production of a BCR/ABL fusion protein. The gene product of the BCR/ABL gene constitutively activates numerous downstream targets including c-myc, Akt and Jun, all of which cause uncontrolled proliferation and survival of CML cells.
Pathogenesis
- The circulating blood cells are produced in bone marrow after a series of events termed as hematopoiesis.[31]
- The bone marrow has an tremendous regenerative ability; it is estimated that 10 trillion red blood cells and 80 to 90 trillion leukocytes are formed per hour at the basal rate.
- In addition to that, while cell numbers are maintained within narrow limits in normal subjects, they can be promptly increased when required.
- Bone marrow primarily has small percentage of pleuripotent stem cells which give rise to various progenitor cells.
- Hematopoeisis occurs in the vertebrae, pelvic bones, metaphysis of long bones such as femur, humerus in basal state.
- However, during certain stressful conditions that require rapid and massive hematopoiesis such as thalassemia it then returns to its former site, liver, spleen and sometimes lymph nodes.
- These hematopoietic stem cells (HSCs are multipotent and have the ability to differentiate into the cells of all 10 blood lineages:
- This differentiation is mediated through multiple growth factors and cytokines. [32] [33]
- The hematopoietic stem cells (HSCs) and progenitor cells are supported by a stromal cell network that provides cell-cell contact support.
- The stromal network provides two major functions:
- An adhesive framework onto which the developing cells are bound, these cells produce:
- A variety of adhesion molecules.
- Hematopoietic Growth factors or cytokines that are thought to support the survival, proliferation, and differentiation of HSCs and progenitors. [34]
- Primitive mesenchymal stromal cells (MSCs) are thought to have the capacity to differentiate into following:
- Osteolineage cells
- Chondrocytes
- Adipocytes
- Perivascular cells
- Overall Differentiation of myeloid progenitors is mediated through:[35]
- The production of essential hematopoietic growth factors.
- Several signaling pathways have come up as integral control devices of HSC fate, such as:[36]
- Notch
- Wingless-type (Wnt)
- Sonic hedgehog (Shh)
- Smad pathways
- These signaling circuits provide an important structure for our understanding of HSC regulation, alongwith providing information of how the bone marrow micro environment couples and integrates extrinsic with intrinsic factors responsible for HSC differentiation and development of chronic myeloid leukemia.[37]
Genetic Translocation:
- Chronic myeloid leukemia (CML), a myeloproliferative neoplasm, characterized by the presence of the Philadelphia chromosome which is thought to be a definitive diagnostic marker for CML.[38]
- In Philadelphia chromosome translocation, parts of two chromosomes (the 9th and 22nd by conventional karyotypic numbering) switch places.
- As a result, part of the BCR ("breakpoint cluster region") gene from chromosome 22 is fused with the ABL ("abelson murine leukemia") gene on chromosome 9.
- This abnormal "fusion" gene generates a protein of p210 .
- Because ABL carries a domain that can add phosphate groups to tyrosine residues (a tyrosine kinase), the BCR/ABL fusion gene product is also a tyrosine kinase.
- The fused BCR/ABL protein interacts with the interleukin 3 beta c receptor subunit.
- The BCR/ABL transcript is continuously active and does not require activation by other cellular messaging proteins that promotes growth and replication through downstream pathways such as:
- RAS
- RAF
- JUN kinase
- MYC
- STAT
- In turn BCR/ABL triggers a cascade of proteins which control the cell cycle, speeding up cell division.
- Moreover the BCR/ABL protein inhibits DNA repair, causing genomic instability and making the cell more susceptible to developing further genetic mutations.
- The action of the BCR/ABL protein is the pathophysiologic cause of chronic myelogenous leukemia.[39][40][41][42][38][39]
Role of reactive oxygen species:
- Recent studies have demonstarated that BCR/ABL also stimulates the production of reactive oxygen species (ROS), which levels increase with CML progression and this in turn increases BCR/ABL self-mutagenesis.
- Tyrosine kinase inhibitor resistance can also be related to higher ROS production.
- Therefore, ROS-induced self-mutagenesis of BCR/ABL is of prime significance for CML progression.
- These can be dependent on DNA repair, which is modulated by BCR/ABL and can be different in CML stem and progenitor cells.[43]
Altered bone marrow pathway signalling:
- Implication of altered bone marrow signalling on stem cell persistence in the bone marrow niche.
- Recent advancements have been trying to establish the relationship between bone marrow pathway and Wnt pathways and their role to alter the Cdx-Hox axi.[44]
Role of Integrin:
- CML cells present in contact with stroma or fibronectin continue to proliferate, suggesting that failure to adhere through integrin receptors may also underlie the abnormal proliferation of CML progenitors.[45]
- Although, CML progenitors express the same integrin receptors as normal progenitors, they fail to adhere to stroma and fibronectin.
- This indicates that structural or functional abnormalities of these receptors can be integral part of pathogenesis.
Blast crisis:
- Chronic myeloid leukemia (CML) in blast crisis is the transition of CML in chronic or accelerated phase to an acute leukemia.
- It is characterized by:
- ≥ 30% blasts in the bone marrow or peripheral blood.
- The development of extramedullary disease outside of the spleen.
- In light of recent changes in the World Health Organization, definition of acute leukemia, the percentage of blasts required for CML in blastic phase may someday be reduced to 20%.[46]
- Consistent with the early stem cell nature of CML, blastic transformation may be:
- Myeloid blast crisis being about two times more common than lymphoid.
- It is suggested that blast crisis is due to one of following reasons:[47]
- BCR/ABL is considered to be responsible for progressive genomic instability or epigenetic changes, which occur at the CML stem cell level and/or in later CML progenitor cells.
- The degree of genomic instability is directly related to the level of BCR/ABL kinase activity.
- The third is that CML stem cells are the least vulnerable to ABL-targeted therapy and may serve as reservoirs for CML progression.
- All these events concomitantly result in the acquired loss of hematopoietic cell differentiation, resulting in a highly progressive generation of immature blasts in peripheral blood and in bone marrow.
- Various studies have BCR/ABL is implicated in the generation and maintenance of secondary DNA alterations.
Genetic Alterations in Blast crisis:
- Following genetic changes have been observed which play crucial role in progression of disease phase.
- Duplication of the Ph chromosome, trisomy 8, and isochromosome 17.[48]
- Alterations in p53 have been found in only a minority of cases.[49]
- Loss of p16INK4A/ARF has been reported in up to half of patients with CML in lymphoid blast crisis but is rare in the myeloid form.[50]
- Thus, it is hypothesized that clonal evolution plays integral role in blastic progression and is likely facilitated by the dysregulation of normal apoptotic pathways by BCR/ABL.
Gross Pathology
On gross pathology, no distinctive findings are seen in chronic myeloid leukemia.
Microscopic Pathology
Blast cells are seen on peripheral blood smear of patients of chronic myeloid leukemia which are present during blast crisis.
-
![Blast crisis of chronic myelogenous leukemia (CML). Peripheral blood smear revealing the histopathologic features indicative of a blast crisis in the case of chronic myelogenous leukemia.[52]](/images/b/ba/CML.jpg)
Blast crisis of chronic myelogenous leukemia (CML). Peripheral blood smear revealing the histopathologic features indicative of a blast crisis in the case of chronic myelogenous leukemia.[52]
![Blast crisis of chronic myelogenous leukemia (CML). Peripheral blood smear revealing the histopathologic features indicative of a blast crisis in the case of chronic myelogenous leukemia.[52]](/index.php/File:CML.jpg)
References
- ↑ Sinclair L (2008). "Recognizing, treating and understanding pernicious anaemia". J R Soc Med. 101 (5): 262–4. doi:10.1258/jrsm.2008.081006. PMC 2376267. PMID 18463283.
- ↑ SMITH EL (1948). "Purification of anti-pernicious anaemia factors from liver". Nature. 161 (4095): 638. doi:10.1038/161638a0. PMID 18856623.
- ↑ Miles LM, Allen E, Clarke R, Mills K, Uauy R, Dangour AD (2017). "Impact of baseline vitamin B12 status on the effect of vitamin B12 supplementation on neurologic function in older people: secondary analysis of data from the OPEN randomised controlled trial". Eur J Clin Nutr. 71 (10): 1166–1172. doi:10.1038/ejcn.2017.7. PMID 28225050.
- ↑ Watanabe F (2007). "Vitamin B12 sources and bioavailability". Exp Biol Med (Maywood). 232 (10): 1266–74. doi:10.3181/0703-MR-67. PMID 17959839.
- ↑ Jung SB, Nagaraja V, Kapur A, Eslick GD (2015). "Association between vitamin B12 deficiency and long-term use of acid-lowering agents: a systematic review and meta-analysis". Intern Med J. 45 (4): 409–16. doi:10.1111/imj.12697. PMID 25583062.
- ↑ 6.0 6.1 Del Corral A, Carmel R (1990). "Transfer of cobalamin from the cobalamin-binding protein of egg yolk to R binder of human saliva and gastric juice". Gastroenterology. 98 (6): 1460–6. doi:10.1016/0016-5085(90)91076-i. PMID 2110915.
- ↑ Harrington DJ (2017). "Laboratory assessment of vitamin B12 status". J Clin Pathol. 70 (2): 168–173. doi:10.1136/jclinpath-2015-203502. PMID 27169753.
- ↑ Tinelli C, Di Pino A, Ficulle E, Marcelli S, Feligioni M (2019). "Hyperhomocysteinemia as a Risk Factor and Potential Nutraceutical Target for Certain Pathologies". Front Nutr. 6: 49. doi:10.3389/fnut.2019.00049. PMC 6491750. PMID 31069230.
- ↑ Callaghan JM, Khan MA, Alderuccio F, van Driel IR, Gleeson PA, Toh BH (1993). "Alpha and beta subunits of the gastric H+/K(+)-ATPase are concordantly targeted by parietal cell autoantibodies associated with autoimmune gastritis". Autoimmunity. 16 (4): 289–95. doi:10.3109/08916939309014648. PMID 7517707.
- ↑ Toh BH, Sentry JW, Alderuccio F (2000). "The causative H+/K+ ATPase antigen in the pathogenesis of autoimmune gastritis". Immunol Today. 21 (7): 348–54. doi:10.1016/s0167-5699(00)01653-4. PMID 10871877.
- ↑ Schade SG, Abels J, Schilling RF (1967). "Studies on antibody to intrinsic factor". J Clin Invest. 46 (4): 615–20. doi:10.1172/JCI105563. PMC 442045. PMID 6021209.
- ↑ Green R, Datta Mitra A (2017). "Megaloblastic Anemias: Nutritional and Other Causes". Med Clin North Am. 101 (2): 297–317. doi:10.1016/j.mcna.2016.09.013. PMID 28189172.
- ↑ Heidelbaugh JJ (2013). "Proton pump inhibitors and risk of vitamin and mineral deficiency: evidence and clinical implications". Ther Adv Drug Saf. 4 (3): 125–33. doi:10.1177/2042098613482484. PMC 4110863. PMID 25083257.
- ↑ Aroda VR, Edelstein SL, Goldberg RB, Knowler WC, Marcovina SM, Orchard TJ; et al. (2016). "Long-term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study". J Clin Endocrinol Metab. 101 (4): 1754–61. doi:10.1210/jc.2015-3754. PMC 4880159. PMID 26900641.
- ↑ Zulfiqar AA, Andres E (2017). "Association pernicious anemia and autoimmune polyendocrinopathy: a retrospective study". J Med Life. 10 (4): 250–253. PMC 5771255. PMID 29362601.
- ↑ Carmel R (1996). "Prevalence of undiagnosed pernicious anemia in the elderly". Arch Intern Med. 156 (10): 1097–100. PMID 8638997.
- ↑ 17.0 17.1 Andres E, Serraj K (2012). "Optimal management of pernicious anemia". J Blood Med. 3: 97–103. doi:10.2147/JBM.S25620. PMC 3441227. PMID 23028239.
- ↑ Gordon MM, Brada N, Remacha A, Badell I, del Río E, Baiget M; et al. (2004). "A genetic polymorphism in the coding region of the gastric intrinsic factor gene (GIF) is associated with congenital intrinsic factor deficiency". Hum Mutat. 23 (1): 85–91. doi:10.1002/humu.10297. PMID 14695536.
- ↑ Cattan D (2011). "Pernicious anemia: what are the actual diagnosis criteria?". World J Gastroenterol. 17 (4): 543–4. doi:10.3748/wjg.v17.i4.543. PMC 3027024. PMID 21274387.
- ↑ Seynabou F, Fatou Samba Diago N, Oulimata Diop D, Abibatou Fall S, Nafissatou D (2016). "Biermer anemia: Hematologic characteristics of 66 patients in a Clinical Hematology Unit at Senegal". Med Sante Trop. 26 (4): 402–407. doi:10.1684/mst.2016.0625. PMID 28073728.
- ↑ Han L, Wu S, Han F, Gu X (2015). "Insights into the molecular mechanisms of methylmalonic acidemia using microarray technology". Int J Clin Exp Med. 8 (6): 8866–79. PMC 4538064. PMID https://www.ncbi.nlm.nih.gov/pubmed/26309541 Check
|pmid=value (help). - ↑ Choi C, Kim T, Park KD, Lim OK, Lee JK (2019). "Subacute Combined Degeneration Caused by Nitrous Oxide Intoxication: A Report of Two Cases". Ann Rehabil Med. 43 (4): 530–534. doi:10.5535/arm.2019.43.4.530. PMC 6734019 Check
|pmc=value (help). PMID 31499607. - ↑ Stoopler ET, Kuperstein AS (2013). "Glossitis secondary to vitamin B12 deficiency anemia". CMAJ. 185 (12): E582. doi:10.1503/cmaj.120970. PMC 3761039. PMID 23359038.
- ↑ Lahner E, Annibale B (2009). "Pernicious anemia: new insights from a gastroenterological point of view". World J Gastroenterol. 15 (41): 5121–8. doi:10.3748/wjg.15.5121. PMC 2773890. PMID 19891010.
- ↑ Korman MG, Strickland RG, Hansky J (1972). "The functional 'G' cell mass in atrophic gastritis". Gut. 13 (5): 349–51. doi:10.1136/gut.13.5.349. PMC 1412218. PMID 5036089.
- ↑ "StatPearls". 2020. PMID 29939561.
- ↑ Farrelly SJ, O'Connor KA (2017). "Hypersegmented neutrophils and oval macrocytes in the setting of B12 deficiency and pancytopaenia". BMJ Case Rep. 2017. doi:10.1136/bcr-2016-218508. PMC 5612428. PMID 28821482.
- ↑ Annibale B, Lahner E, Fave GD (2011). "Diagnosis and management of pernicious anemia". Curr Gastroenterol Rep. 13 (6): 518–24. doi:10.1007/s11894-011-0225-5. PMID 21947876.
- ↑ Murphy G, Dawsey SM, Engels EA, Ricker W, Parsons R, Etemadi A; et al. (2015). "Cancer Risk After Pernicious Anemia in the US Elderly Population". Clin Gastroenterol Hepatol. 13 (13): 2282-9.e1-4. doi:10.1016/j.cgh.2015.05.040. PMC 4655146. PMID 26079040.
- ↑ Venerito M, Link A, Rokkas T, Malfertheiner P (2016). "Gastric cancer - clinical and epidemiological aspects". Helicobacter. 21 Suppl 1: 39–44. doi:10.1111/hel.12339. PMID 27531538.
- ↑ Blank U, Karlsson G, Karlsson S (January 2008). "Signaling pathways governing stem-cell fate". Blood. 111 (2): 492–503. doi:10.1182/blood-2007-07-075168. PMID 17914027.
- ↑ Wilson A, Trumpp A (February 2006). "Bone-marrow haematopoietic-stem-cell niches". Nat. Rev. Immunol. 6 (2): 93–106. doi:10.1038/nri1779. PMID 16491134.
- ↑ Blank U, Karlsson G, Karlsson S (January 2008). "Signaling pathways governing stem-cell fate". Blood. 111 (2): 492–503. doi:10.1182/blood-2007-07-075168. PMID 17914027.
- ↑ Smith C (2003). "Hematopoietic stem cells and hematopoiesis". Cancer Control. 10 (1): 9–16. doi:10.1177/107327480301000103. PMID 12598852.
- ↑ Chereda B, Melo JV (April 2015). "Natural course and biology of CML". Ann. Hematol. 94 Suppl 2: S107–21. doi:10.1007/s00277-015-2325-z. PMID 25814077.
- ↑ Blank U, Karlsson G, Karlsson S (January 2008). "Signaling pathways governing stem-cell fate". Blood. 111 (2): 492–503. doi:10.1182/blood-2007-07-075168. PMID 17914027.
- ↑ Smith C (2003). "Hematopoietic stem cells and hematopoiesis". Cancer Control. 10 (1): 9–16. doi:10.1177/107327480301000103. PMID 12598852.
- ↑ 38.0 38.1 Thompson PA, Kantarjian HM, Cortes JE (October 2015). "Diagnosis and Treatment of Chronic Myeloid Leukemia in 2015". Mayo Clin. Proc. 90 (10): 1440–54. doi:10.1016/j.mayocp.2015.08.010. PMC 5656269. PMID 26434969.
- ↑ 39.0 39.1 Jabbour E, Parikh SA, Kantarjian H, Cortes J (October 2011). "Chronic myeloid leukemia: mechanisms of resistance and treatment". Hematol. Oncol. Clin. North Am. 25 (5): 981–95, v. doi:10.1016/j.hoc.2011.09.004. PMC 4428141. PMID 22054730.
- ↑ Hehlmann R, Hochhaus A, Baccarani M; European LeukemiaNet (2007). "Chronic myeloid leukaemia". Lancet. 370 (9584): 342–50. PMID 17662883.
- ↑ Jabbour E, Kantarjian H (May 2014). "Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management". Am. J. Hematol. 89 (5): 547–56. doi:10.1002/ajh.23691. PMID 24729196.
- ↑ Kaleem B, Shahab S, Ahmed N, Shamsi TS (2015). "Chronic Myeloid Leukemia--Prognostic Value of Mutations". Asian Pac. J. Cancer Prev. 16 (17): 7415–23. PMID 26625737.
- ↑ Antoszewska-Smith J, Pawlowska E, Blasiak J (2017). "Reactive oxygen species in BCR-ABL1-expressing cells - relevance to chronic myeloid leukemia". Acta Biochim. Pol. 64 (1): 1–10. doi:10.18388/abp.2016_1396. PMID 27904889.
- ↑ Toofan P, Wheadon H (October 2016). "Role of the bone morphogenic protein pathway in developmental haemopoiesis and leukaemogenesis". Biochem. Soc. Trans. 44 (5): 1455–1463. doi:10.1042/BST20160104. PMID 27911727.
- ↑ Verfaillie, Catherine M.; Hurley, Randolph; Zhao, Robert C.H.; Prosper, Felipe; Delforge, Michel; Bhatia, Ravi (1997). "Pathophysiology of CML: Do defects in integrin function contribute to the premature circulation and massive expansion of the BCR/ABL positive clone?". Journal of Laboratory and Clinical Medicine. 129 (6): 584–591. doi:10.1016/S0022-2143(97)90192-X. ISSN 0022-2143.
- ↑ Martin PJ, Najfeld V, Hansen JA, Penfold GK, Jacobson RJ, Fialkow PJ (September 1980). "Involvement of the B-lymphoid system in chronic myelogenous leukaemia". Nature. 287 (5777): 49–50. PMID 6968038.
- ↑ Salloukh HF, Laneuville P (August 2000). "Increase in mutant frequencies in mice expressing the BCR-ABL activated tyrosine kinase". Leukemia. 14 (8): 1401–4. PMID 10942235.
- ↑ Kantarjian HM, Keating MJ, Talpaz M, Walters RS, Smith TL, Cork A, McCredie KB, Freireich EJ (September 1987). "Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients". Am. J. Med. 83 (3): 445–54. PMID 3477958.
- ↑ Ahuja, H.; Bar-Eli, M.; Advani, S. H.; Benchimol, S.; Cline, M. J. (1989). "Alterations in the p53 gene and the clonal evolution of the blast crisis of chronic myelocytic leukemia". Proceedings of the National Academy of Sciences. 86 (17): 6783–6787. doi:10.1073/pnas.86.17.6783. ISSN 0027-8424.
- ↑ Hasford J, Pfirrmann M, Hehlmann R, Baccarani M, Guilhot F, Mahon FX, Kluin-Nelemans HC, Ohnishi K, Thaler J, Steegmann JL (January 2003). "Prognosis and prognostic factors for patients with chronic myeloid leukemia: nontransplant therapy". Semin. Hematol. 40 (1): 4–12. doi:10.1053/shem.2003.50006. PMID 12563607.
- ↑ Donato, N. J. (2003). "BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571". Blood. 101 (2): 690–698. doi:10.1182/blood.V101.2.690. ISSN 0006-4971.
- ↑ Center for Disease Control and Prevention. Public Health Image Library 2015.http://phil.cdc.gov/phil/details_linked.asp?pid=6