Atherosclerosis pathophysiology
|
Atherosclerosis Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
ACC/AHA Guideline Recommendations |
|
Case Studies |
|
Atherosclerosis pathophysiology On the Web |
|
American Roentgen Ray Society Images of Atherosclerosis pathophysiology |
|
Risk calculators and risk factors for Atherosclerosis pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]Assistant Editor-in-Chief: Mohammad I. Barouqa, M.D.[2]
Synonyms and keywords: Atherosclerosis, Atherogenesis, Atheroma, Coronary Artery Disease, CAD, Cerebrovascular Disease
Overview
Atherothrombotic diseases are a major healthcare dilemma and contribute to more that 25% of all deaths worldwide.Recent WHO reports indicate that the disease affect developing and developed countries making atherothrombotic diseases a global challenge.[1]
Atherosclerosis is a chronic inflammatory process of the arterial wall which involves an imbalanced lipid metabolism and a maladaptive immune response. The abnormal process of lipid accumulation, immune responses and their clearance is controlled by leukocytes and homeostasis which act in response to chemokines.
Atherogenesis is the developmental process of atheromatous plaques. It is characterized by a remodeling of arteries involving the concomitant accumulation of fatty substances called plaques. One recent theory suggests that for unknown reasons, leukocytes such as monocytes or basophils begin to attack the endothelium of the artery lumen in cardiac muscle. The ensuing inflammation leads to formation of atheromatous plaques in the arterial tunica intima, a region of the vessel wall located between the endothelium and the tunica media and tunica adventitia. The bulk of these lesions are made of excess fat, collagen, and elastin. Initially, as the plaques grow only wall thickening occurs without any narrowing, stenosis of the artery opening, called the lumen; stenosis is a late event which may never occur and is often the result of repeated plaque rupture and healing responses, not the just atherosclerosis process by itself.
Pathophysiology
Pathologically, the atheromatous plaque is divided into three distinct components:
- The atheroma ("lump of porridge", from Athera, porridge in Greek,) is the nodular accumulation of a soft, flaky, yellowish material at the center of large plaques, composed of macrophages nearest the lumen of the artery.
- Underlying areas of cholesterol crystals.
- Calcification at the outer base of older/more advanced lesions.
Cellular
The first phase of atherogenesis is the development of fatty streaks. Histological studies showed that focal thickening of the intima with an increase in smooth muscle cells and extracellular matrix initiate the formation of fatty streaks.[2] These smooth muscle cells,some of which originate from hematopoietic stem cells, migrate and proliferate within the intima.Lipids accumulate early in fatty streaks and lead to the formation of both intracellular and extracellular lipid deposits.Among the lipids deposited is the Biglycan,a small dermatan sulfate proteoglycan,which can trap and bind Apolipoprotein E,VLDL remnants,LDL and HDL.[3]Fatty streaks contain Macrophages and variable amounts of T-Lymphocytes which release a variety of inflammatory mediators.When these Macrophages are filled with fats within fatty streaks,they turn into foam cells.These foam cells are considered to be the hallmark of early atheroma.
As fatty streaks expand, more smooth muscle cells migrate to intima and more lipid deposits will be formed. The smooth muscle cells within the deep layers of fatty streaks undergo apoptosis. Once apoptosis occur, more macrophages migrate along with microvesicles and calcify later on. This process contributes to the transition of fatty streaks into atherosclerotic plagues.[4]
LDL in blood plasma poses a risk for cardiovascular disease when it invades the endothelium and becomes oxidized. A complex set of biochemical reactions regulates the oxidation of LDL, chiefly stimulated by presence of free radicals in the endothelium or blood vessel lining.
The initial damage to the blood vessel wall results in a "call for help," an inflammation response. Monocytes (a type of white blood cell) enter the artery wall from the bloodstream, with platelets adhering to the area of insult. This may be promoted by redox signaling induction of factors such as VCAM-1, which recruit circulating monocytes. The monocytes differentiate into macrophages, which ingest oxidized LDL, slowly turning into large "foam cells" – so-described because of their changed appearance resulting from the numerous internal cytoplasmic vesicles and resulting high lipid content.It is worth to note that the oxidation of LDL is mandatory for their uptake by macrophages via unregulated macrophage sacavenger receptors. Under the microscope, the lesion now appears as a fatty streak.Although the LDL uptake may initially be an adaptive process, which prevents LDL-induced endothelial damage, this sort of accumulation results in mitochondrial dysfunction ,apoptosis and necrosis. Thus,Foam cells eventually die, and further propagate the inflammatory process via the release of proteases, inflammatory cytokines and prothrombotic molecules. There is also smooth muscle proliferation and migration from tunica media to intima responding to cytokines secreted by damaged endothelial cells. This would cause the formation of a fibrous capsule covering the fatty streak.
Calcification and Lipids
Intracellular microcalcifications form within vascular smooth muscle cells of the surrounding muscular layer, specifically in the muscle cells adjacent to the atheromas. In time, as cells die, this leads to extracellular calcium deposits between the muscular wall and outer portion of the atheromatous plaques.
Cholesterol is delivered into the vessel wall by cholesterol-containing low-density lipoprotein (LDL) particles. To attract and stimulate macrophages, the cholesterol must be released from the LDL particles and oxidized, a key step in the ongoing inflammatory process. The process is worsened if there is insufficient high-density lipoprotein (HDL), the lipoprotein particle that removes cholesterol from tissues and carries it back to the liver.
The foam cells and platelets encourage the migration and proliferation of smooth muscle cells, which in turn ingest lipids, become replaced by collagen and transform into foam cells themselves. A protective fibrous cap normally forms between the fatty deposits and the artery lining (the intima).
These capped fatty deposits (now called atheromas) produce enzymes that cause the artery to enlarge over time. As long as the artery enlarges sufficiently to compensate for the extra thickness of the atheroma, then no narrowing, stenosis, of the opening, lumen, occurs. The artery becomes expanded with an egg-shaped cross-section, still with a circular opening. If the enlargement is beyond proportion to the atheroma thickness, then an aneurysm is created.[5]
Visible Features
Although arteries are not typically studied microscopically, two plaque types can be distinguished[3]:
- The fibro-lipid (fibro-fatty) plaque is characterized by an accumulation of lipid-laden cells underneath the intima of the arteries, typically without narrowing the lumen due to compensatory expansion of the bounding muscular layer of the artery wall. Beneath the endothelium there is a "fibrous cap" covering the atheromatous "core" of the plaque. The core consists of lipid-laden cells (macrophages and smooth muscle cells) with elevated tissue cholesterol and cholesterol ester content, fibrin, proteoglycans, collagen, elastin and cellular debris. In advanced plaques, the central core of the plaque usually contains extracellular cholesterol deposits (released from dead cells), which form areas of cholesterol crystals with empty, needle-like clefts. At the periphery of the plaque are younger "foamy" cells and capillaries. These plaques usually produce the most damage to the individual when they rupture.
- The fibrous plaque is also localized under the intima, within the wall of the artery resulting in thickening and expansion of the wall and, sometimes, spotty localized narrowing of the lumen with some atrophy of the muscular layer. The fibrous plaque contains collagen fibres (eosinophilic), precipitates of calcium (hematoxylinophilic) and, rarely, lipid-laden cells.
In effect, the muscular portion of the artery wall forms small aneurysms just large enough to hold the atheroma that are present. The muscular portion of artery walls usually remain strong, even after they have remodeled to compensate for the atheromatous plaques.
However, atheromas within the vessel wall are soft and fragile with little elasticity. Arteries constantly expand and contract with each heartbeat, i.e., the pulse. In addition, the calcification deposits between the outer portion of the atheroma and the muscular wall, as they progress, lead to a loss of elasticity and stiffening of the artery as a whole.
The calcification deposits, after they have become sufficiently advanced, are partially visible on coronary artery computed tomography or electron beam tomography (EBT) as rings of increased radiographic density, forming halos around the outer edges of the atheromatous plaques, within the artery wall. On CT, >130 units on the Hounsfield scale (some argue for 90 units) has been the radiographic density usually accepted as clearly representing tissue calcification within arteries. These deposits demonstrate unequivocal evidence of the disease, relatively advanced, even though the lumen of the artery is often still normal by angiographic or intravascular ultrasound.
-
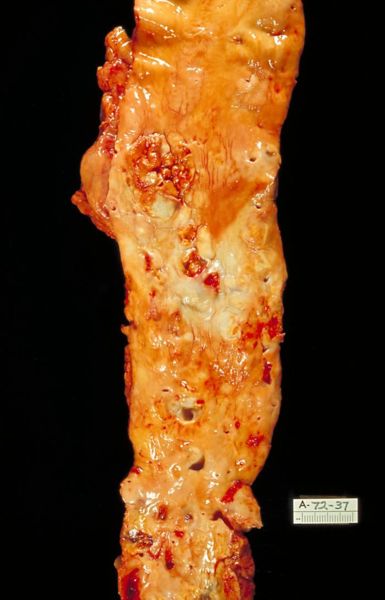
-
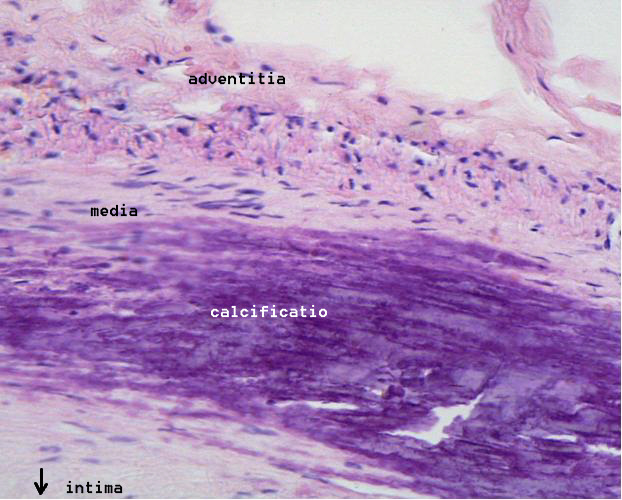
Microphotography of arterial wall with calcified (violet colour) atherosclerotic plaque (haematoxillin & eosin stain)


Rupture and Stenosis
Although the disease process tends to be slowly progressive over decades, it usually remains asymptomatic until an atheroma obstructs the bloodstream in the artery. This is typically by rupture of an atheroma, clotting and fibrous organization of the clot within the lumen, covering the rupture but also producing stenosis, or over time and after repeated ruptures, resulting in a persistent, usually localized stenosis. Stenoses can be slowly progressive, while plaque rupture is a sudden event that occurs specifically in atheromas with thinner/weaker fibrous caps that have become "unstable".
Repeated plaque ruptures, ones not resulting in total lumen closure, combined with the clot patch over the rupture and healing response to stabilize the clot, is the process that produces most stenoses over time. The stenotic areas tend to become more stable, despite increased flow velocities at these narrowings. Most major blood-flow-stopping events occur at large plaques, which, prior to their rupture, produced very little if any stenosis.
From clinical trials, 20% is the average stenosis at plaques that subsequently rupture with resulting complete artery closure. Most severe clinical events do not occur at plaques that produce high-grade stenosis. From clinical trials, only 14% of heart attacks occur from artery closure at plaques producing a 75% or greater stenosis prior to the vessel closing.
If the fibrous cap separating a soft atheroma from the bloodstream within the artery ruptures, tissue fragments are exposed and released, and blood enters the atheroma within the wall and sometimes results in a sudden expansion of the atheroma size. Tissue fragments are very clot-promoting, containing collagen and tissue factor; they activate platelets and activate the system of coagulation. The result is the formation of a thrombus (blood clot) overlying the atheroma, which obstructs blood flow acutely. With the obstruction of blood flow, downstream tissues are starved of oxygen and nutrients. If this is the myocardium (heart muscle), angina (cardiac chest pain) or myocardial infarction (heart attack) develops.
Rather than a single plaque rupture, atherosclerosis is the result of multiple plaque ruptures over time. In patients with ACS, autopsy data show that there had been 5-7 ruptures before the terminal event. These events often due to not lead to a coronary event. Thin capped fibroatheroma (TCFA) with rupture is the most common form of plaque rupture. A less common form is erosion of the plaque. Intraplaque hemorrhage as a result of rupture of the vasa vasorum may also occur. Most ACS is due to TCFA plaque rupture. Less likely there is plaque erosion in ACS. Younger women more often have plaque erosion. Older women more often have plaque rupture. Plaque rupture is usually the mechanism in young men. In older men, the mechanism is similar to women. You cannot predict what plaque is going to rupture on the angiogram. Virtual histology and OCT may potentially predict plaque rupture, but the odds of rupture are so low that their utility is marginal.
Pathological Findings
-
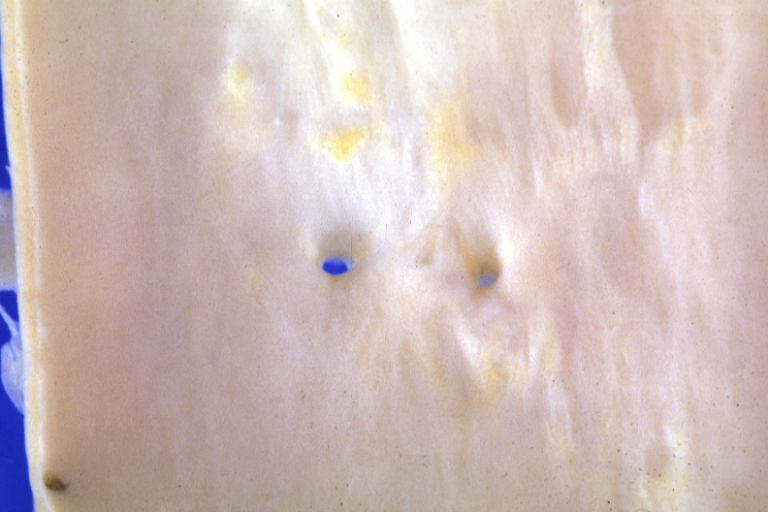
Atherosclerosis: Gross, close-up of fatty streak and intimal thickening
-
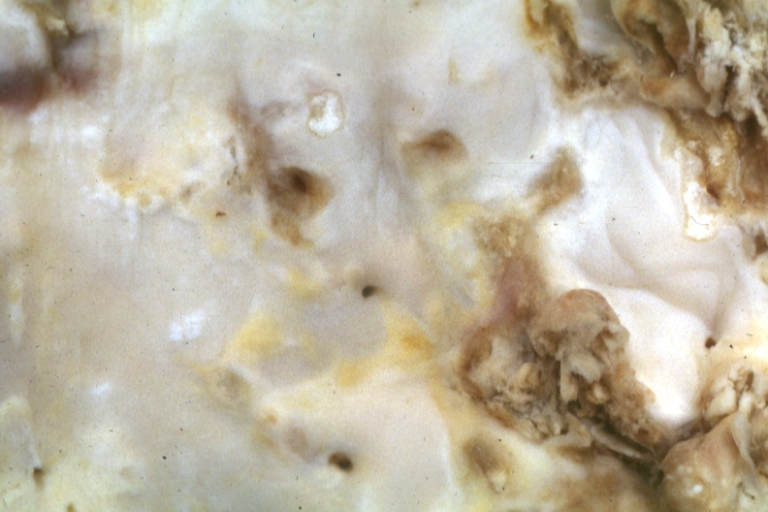
Atherosclerosis: Gross, very good example of fibrous plaques with ulceration and thrombosis
-
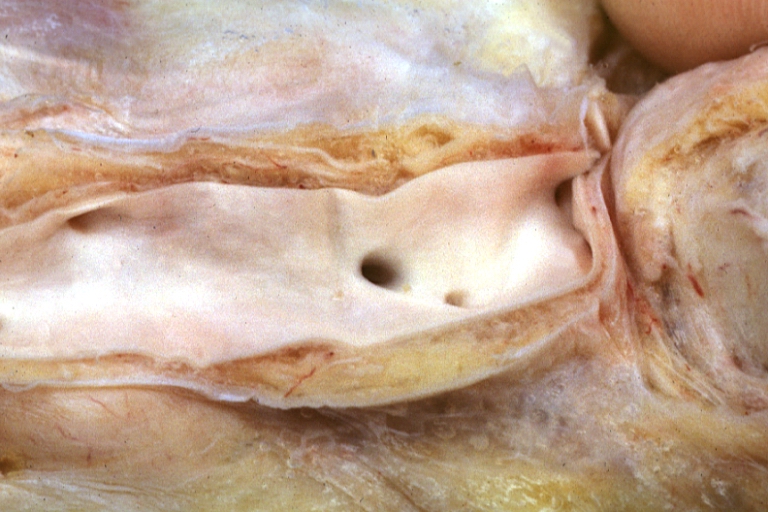
Atherosclerosis: Gross, proximal left anterior descending artery showing faint fatty streaks and penetrating arteries
-
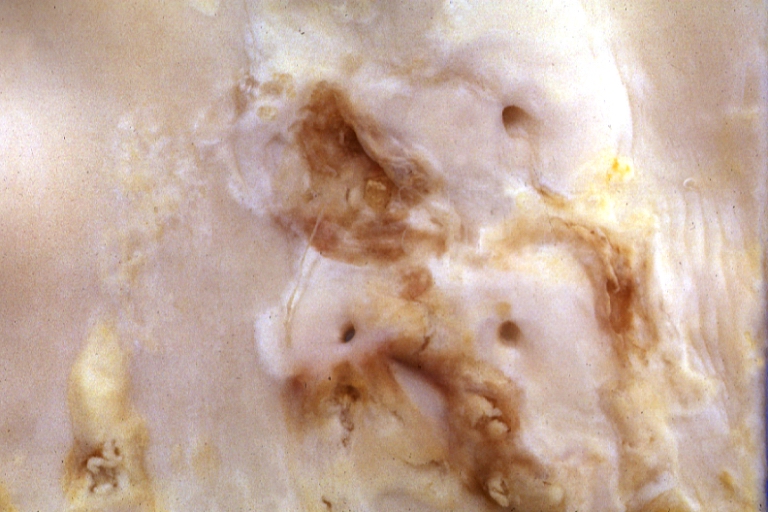
Atherosclerosis: Gross, close-up view of aorta. A plaque with ulceration and thrombosis




-
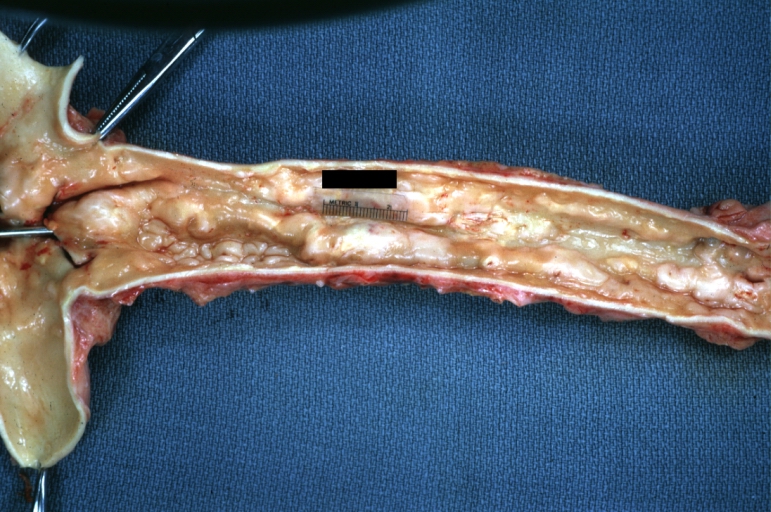
Atherosclerosis: Gross, good example of plaques in aorta
-
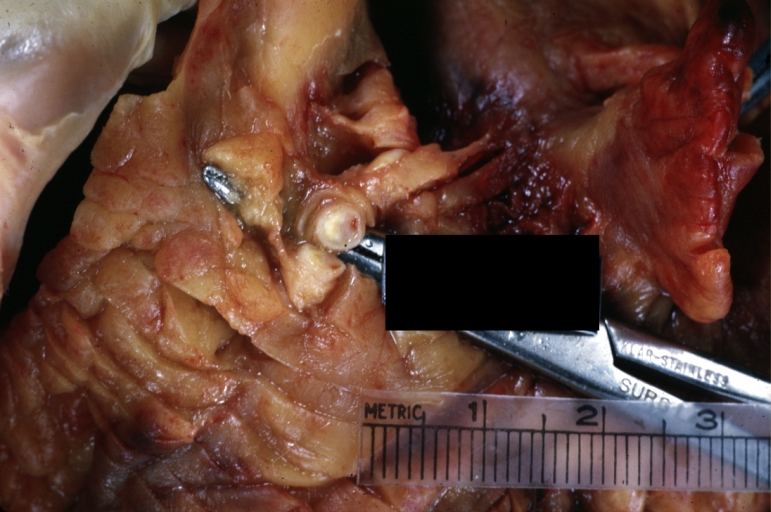
Atherosclerosis: Coronary artery: Gross, close-up view of excellent plaque lesion causing more than 90% occlusion
-
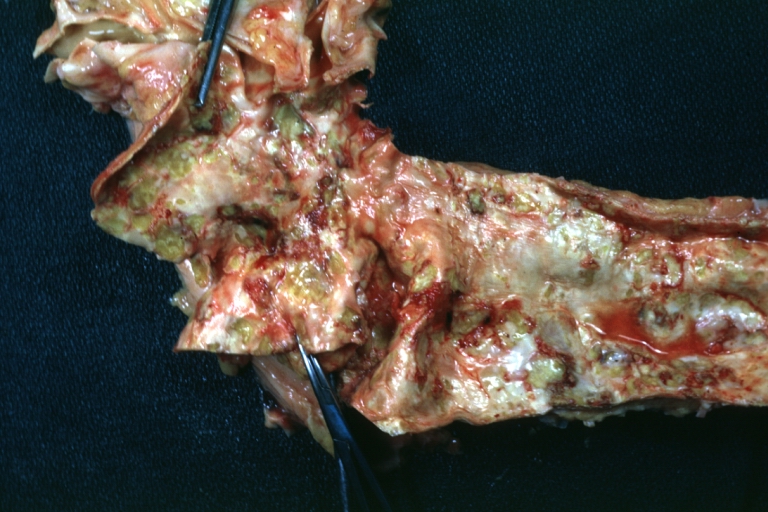
Atherosclerosis: Gross, good example of advanced calcific atherosclerosis in aorta.
-
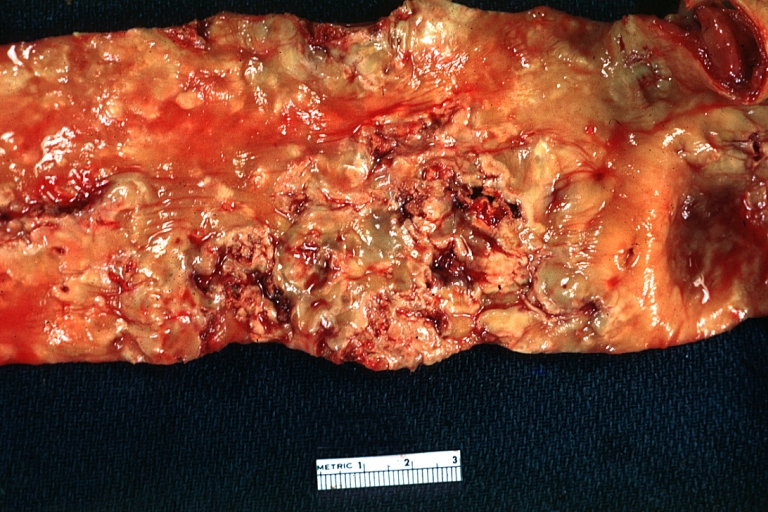
Atherosclerosis: Gross, very good example of calcified and ulcerated atheromatous plaques in aorta




-
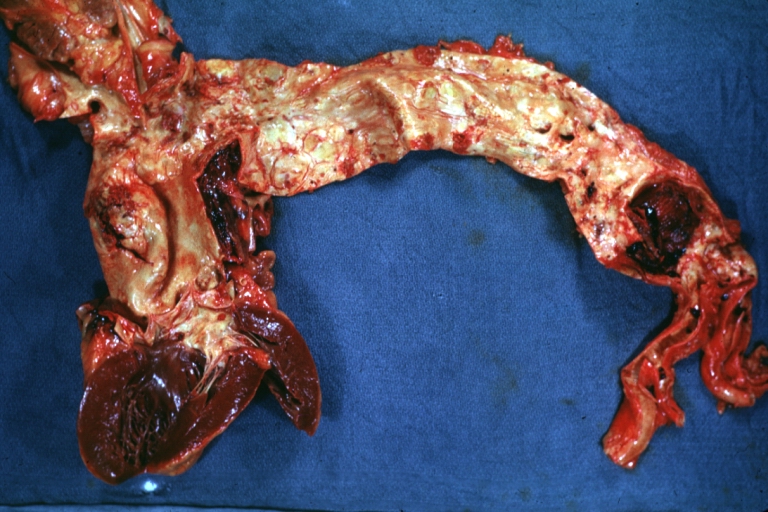
Atherosclerosis: Dissecting Aortic Aneurysm: Gross, shows dilated aorta with extensive atherosclerosis dissection is seen. A small abdominal aorta, atherosclerotic aneurysm is present. A good picture for association of dilation with dissection
-
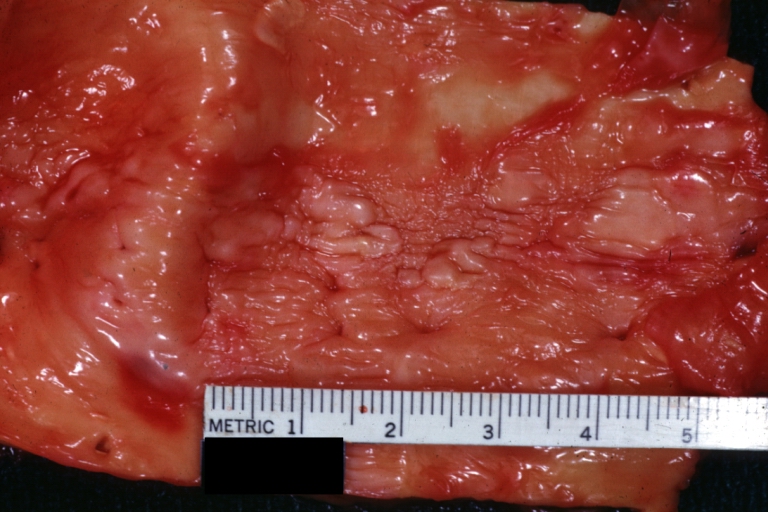
Atherosclerosis: Gross, close-up, an excellent view but appearance much like that of syphilitic aortitis
-
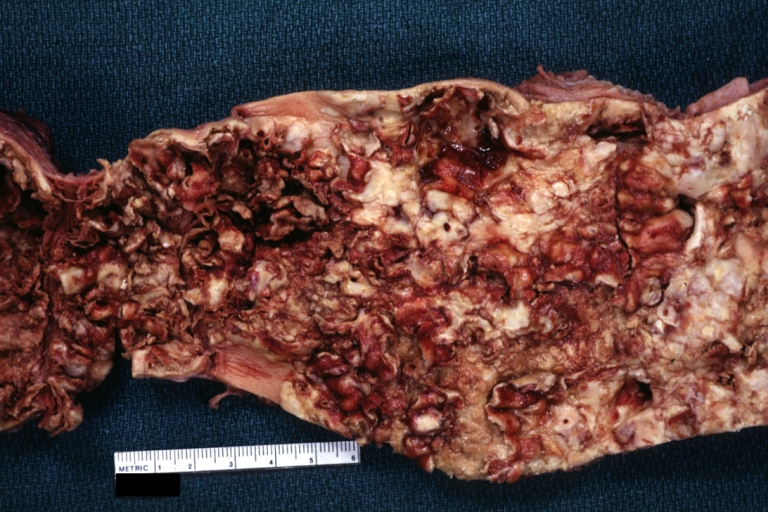
Atherosclerosis: Gross, an excellent example of ulcerated lesions with many mural thrombi
-
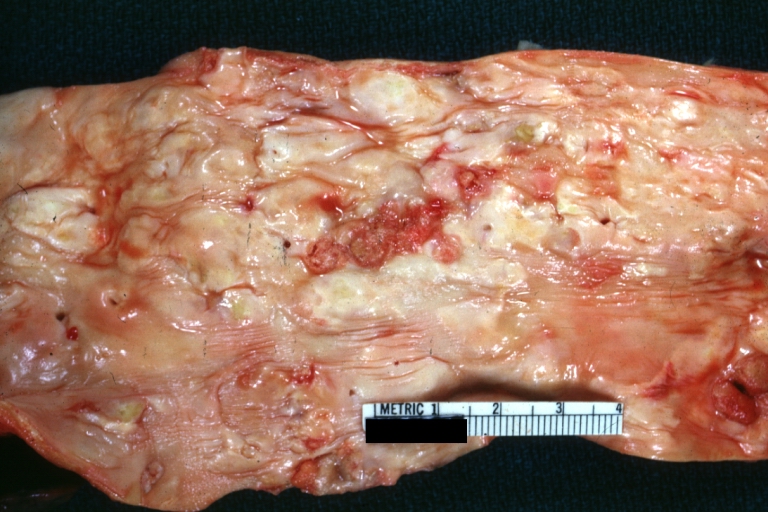
Atherosclerosis: Gross, very good example of plaque lesion and small mural thrombi




-
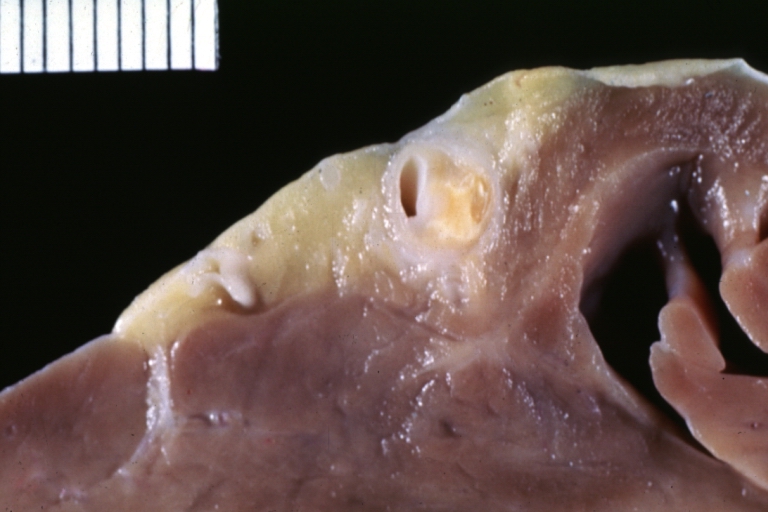
Atherosclerosis: Coronary artery: Gross, an excellent example of plaque, 80% occlusion, uncomplicated lesion.
-
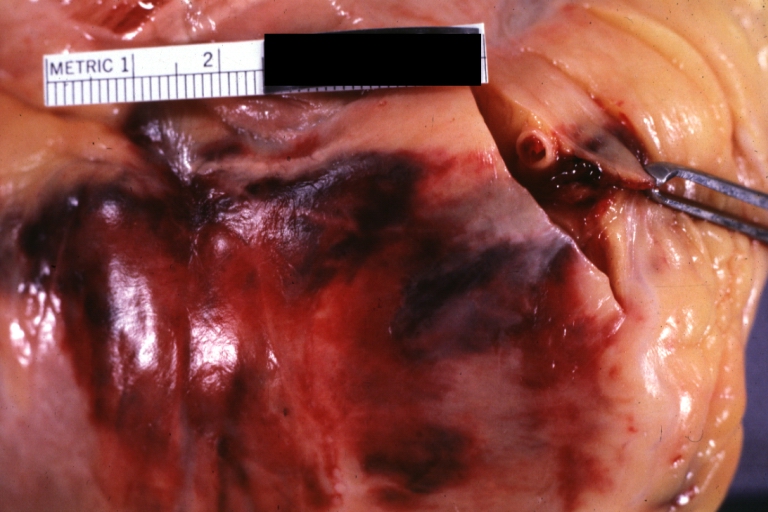
Atherosclerosis: Coronary artery: Atherosclerosis and thrombotic occlusion: Gross, (an excellent example) in situ on heart
-
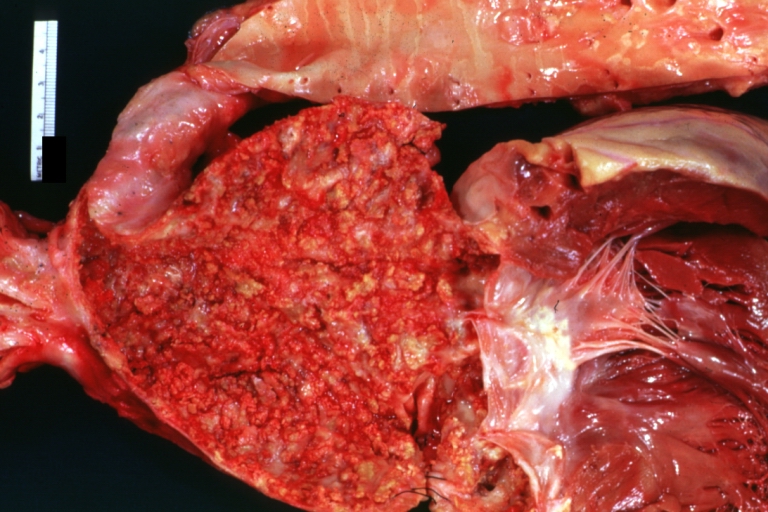
Atherosclerosis: Coarctation: Gross, adult lesion with dramatic demonstration of accelerated atherosclerosis
-
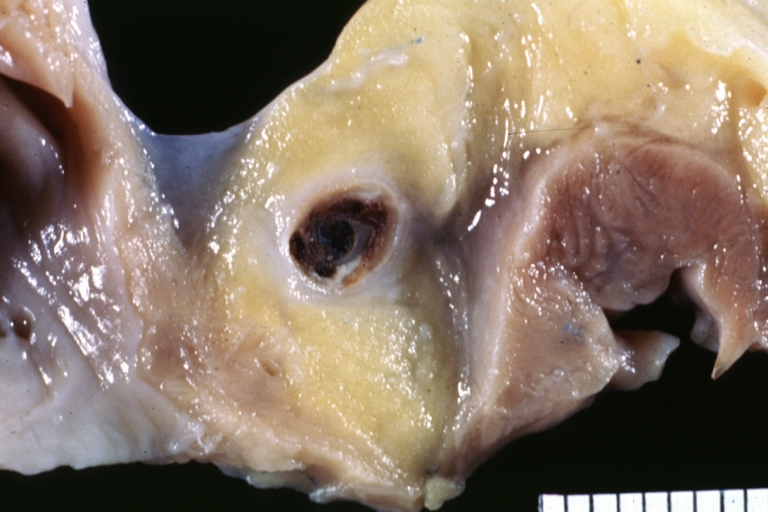
Atherosclerosis: Coronary artery: Gross, cross section well shown hemorrhage into plaque and thrombosis




-
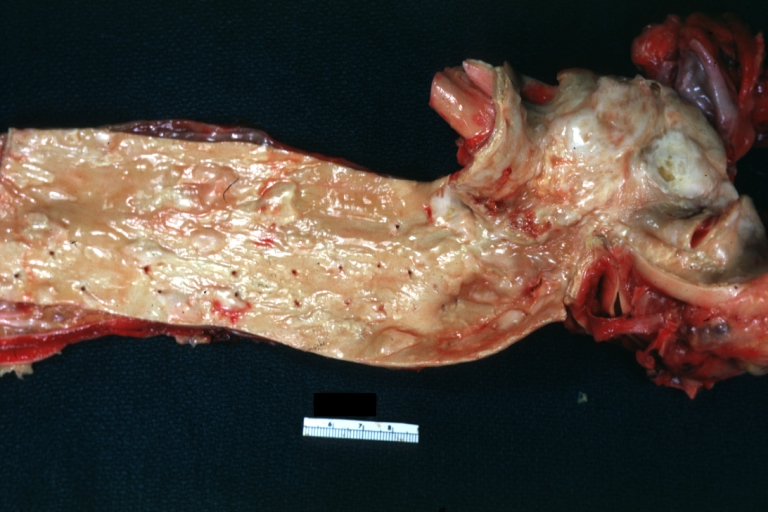
Atherosclerosis: Aorta: Gross, good example of fibrous plaques
-
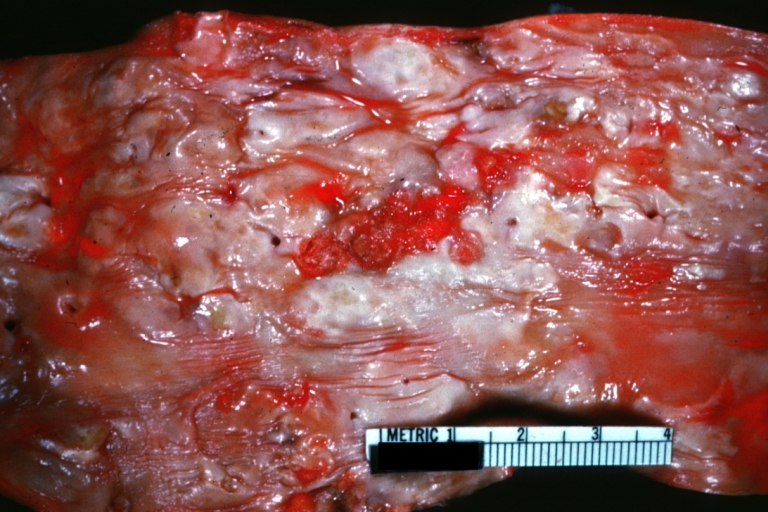
Atherosclerosis: Aorta: Gross, an excellent example of fibrous plaques and mural thrombi
-
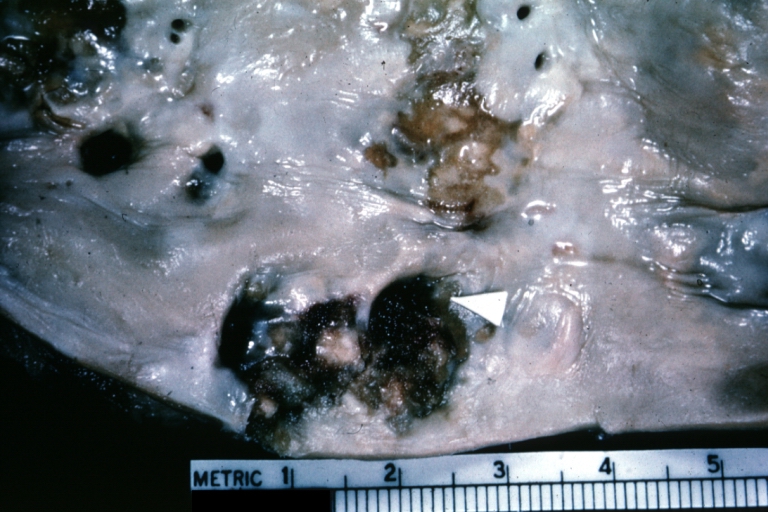
Atherosclerosis: Aorta: Gross, thrombi at the origin of celiac axis and superior mesenteric arteries
-
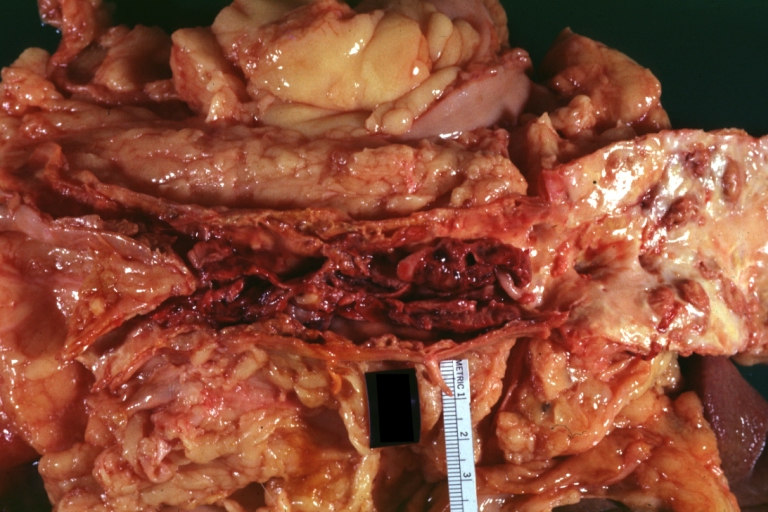
Atherosclerosis: Aorta: Gross, thrombotic occlusion extending from just below renal arteries into iliac artery




-
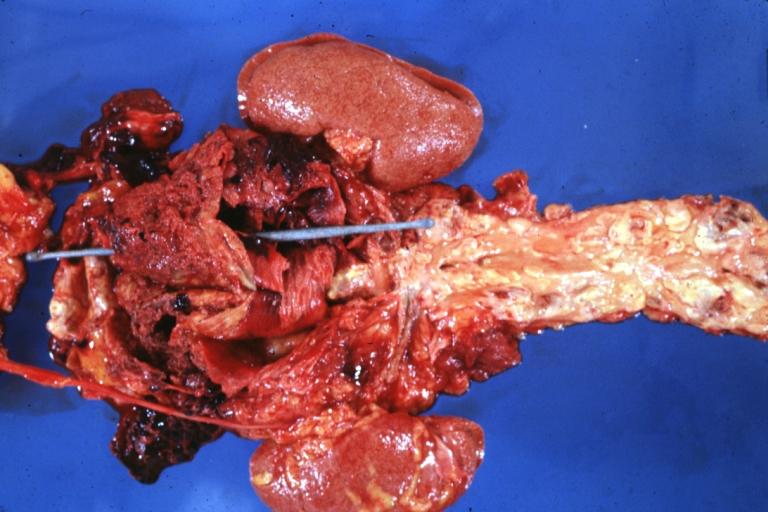
Atherosclerosis: Abdominal Aneurysm Ruptured: Gross, a good example, opened kidneys in marked place. Atherosclerosis in lower thoracic aorta
-
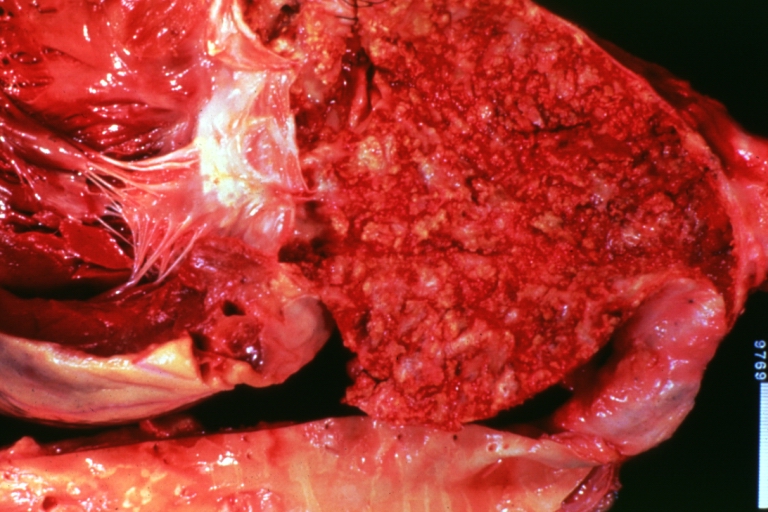
Atherosclerosis: Adult type coarctation with atherosclerosis in aortic arch
-
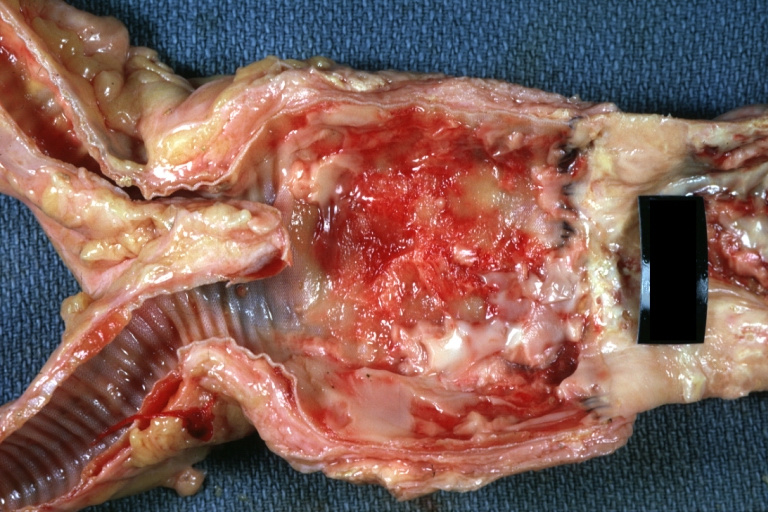
Atherosclerosis: Abdominal Aneurysm Graft Repair: Gross natural color, close-up, an excellent example of Dacron graft that has been in place for years with pseudointima and atherosclerosis
-
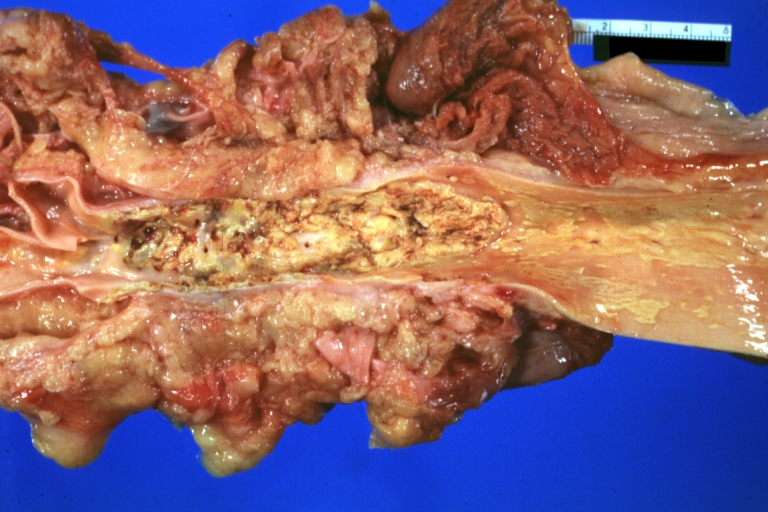
Atherosclerosis: Renal Transplant: Gross, natural color, a close-up view of severe atherosclerosis in abdominal segment of aorta and fatty streaks in descending thoracic much advanced lesions for a 22 years old male with chronic glomerulonephritis




Saphenous Vein Graft
-
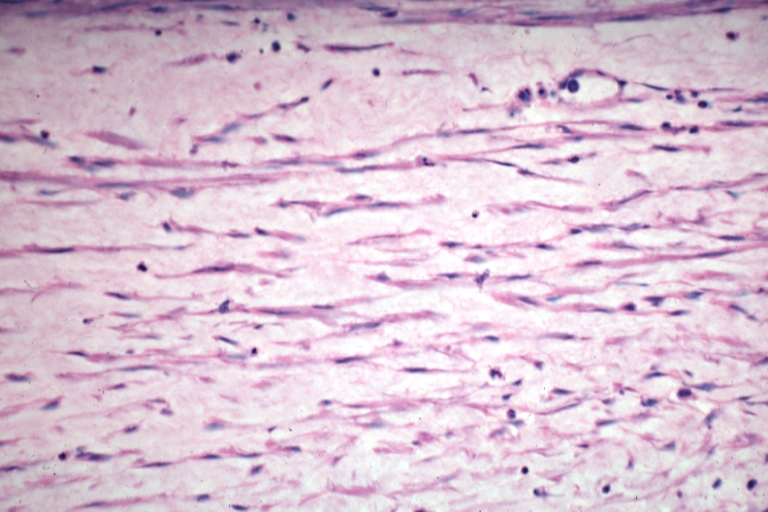
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, an excellent demonstration of myofibroblastic type cells in thickened intima
-
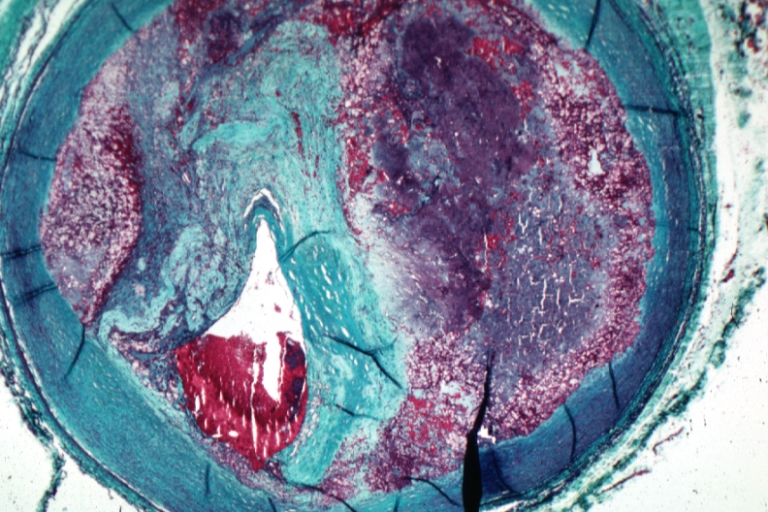
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, trichrome low mag, advanced plaque with hemorrhage into atheroma and complete lumen occlusion with fresh thrombus
-
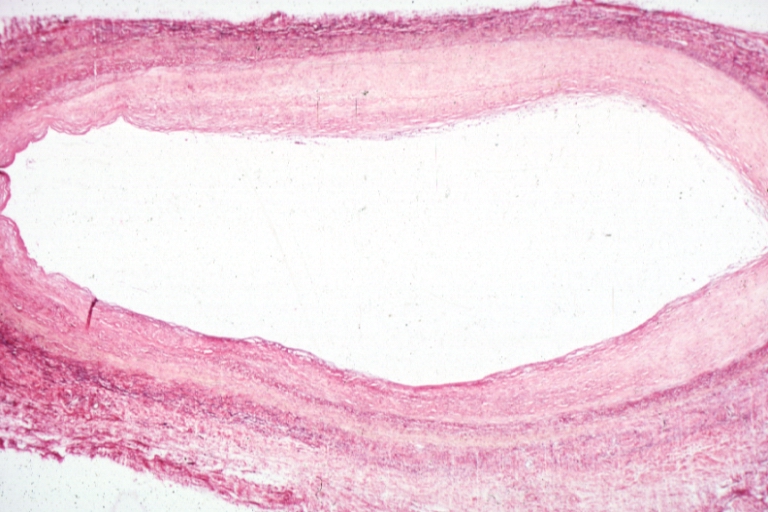
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, low mag, early atheromata consisting of subendothelial foam cells, whole graft and fibrous intimal thickening, (102 months after CABG)
-
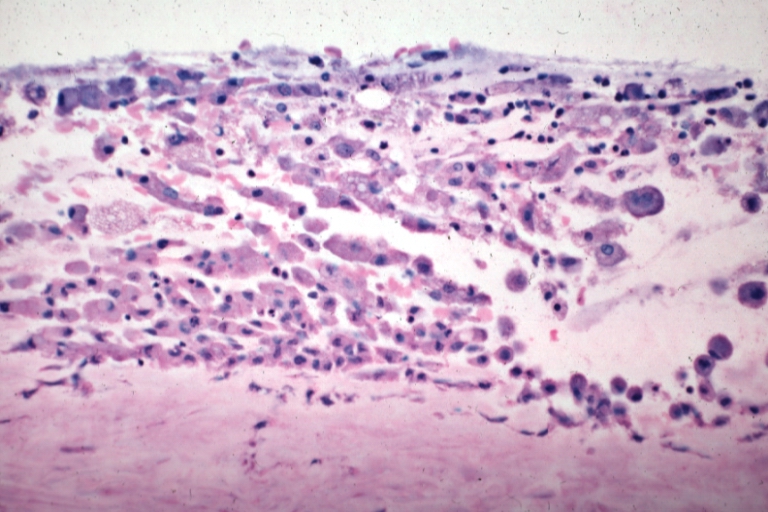
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, high mag, subendothelial foam cells




-
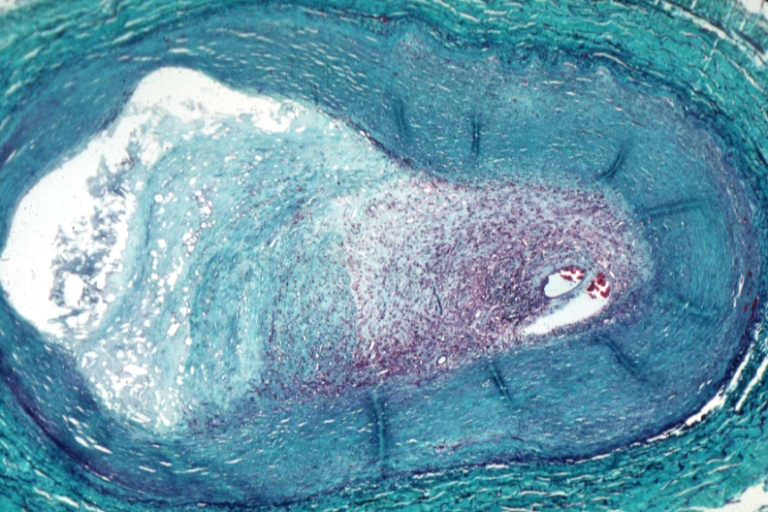
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, trichrome, low mag, large athero plaque and recanalized thrombus in lumen
-
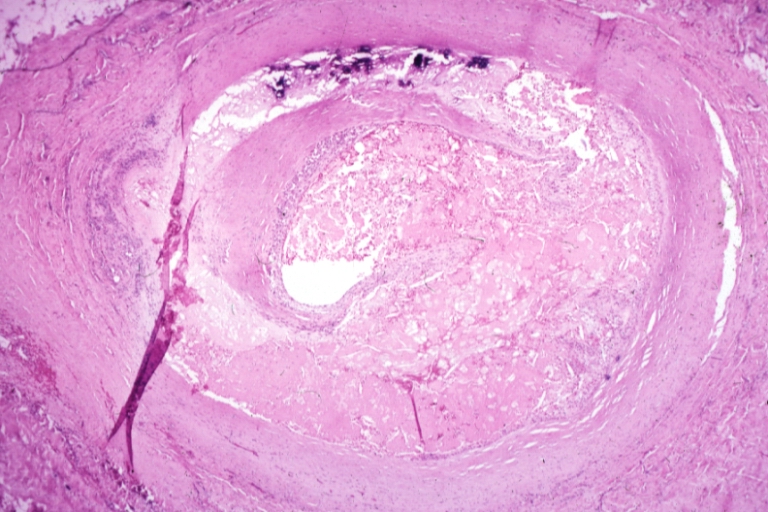
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, H&E, low mag, large and ruptured atheromatous plaque with focal hemorrhage and calcification
-
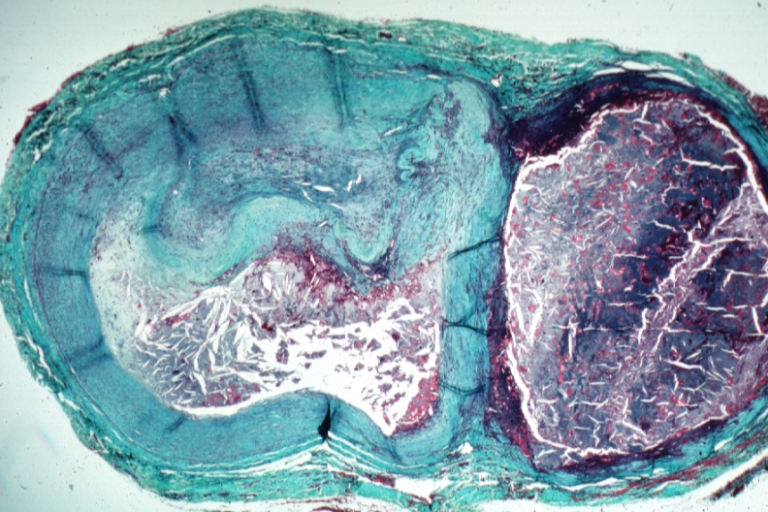
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, trichrome, low mag, very complicated hemorrhagic and necrotic plaque with lumen occlusion. A recanalized thrombus and a large aneurysm at side filled with old blood and atheroma crystals
-
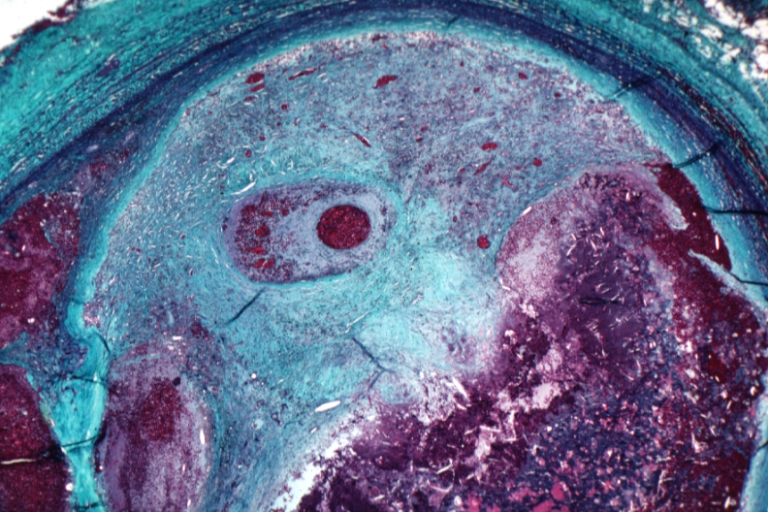
Saphenous vein coronary bypass graft: Atherosclerosis: Micro, ald. fusch., complicated occlusive lesion with organized and recanalized thrombus and old hemorrhage into atheroma




Coronary Arteries
-
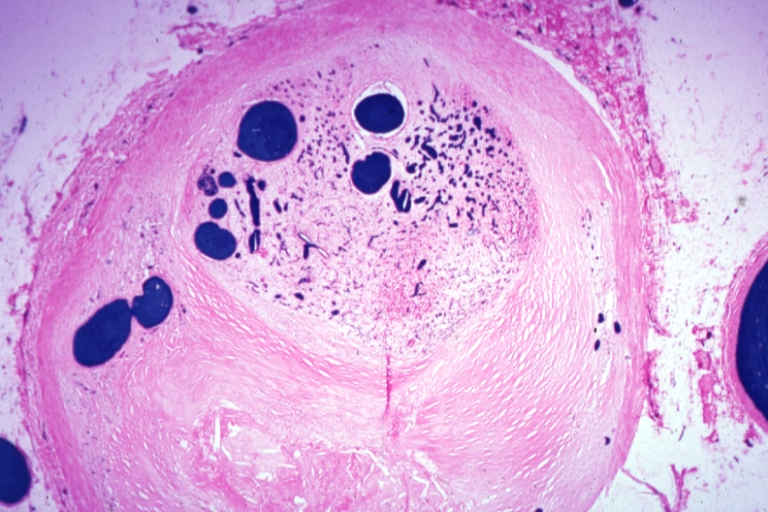
Coronary artery: Atherosclerosis: Micro, H&E, low mag, injected artery, good demonstration of organized and recanalized thrombus
-
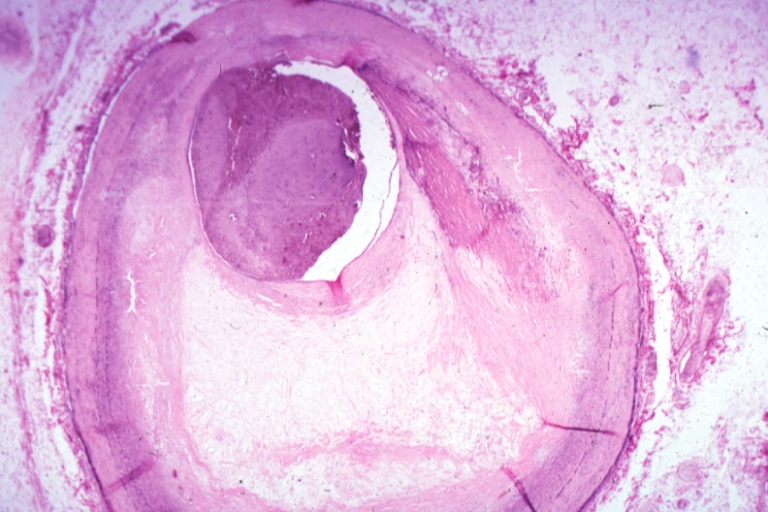
Coronary artery: Atherosclerosis: Micro, H&E, low mag, injected artery, very good example of marked lumen stenosis due to typical fibrous plaque with some calcifications
-
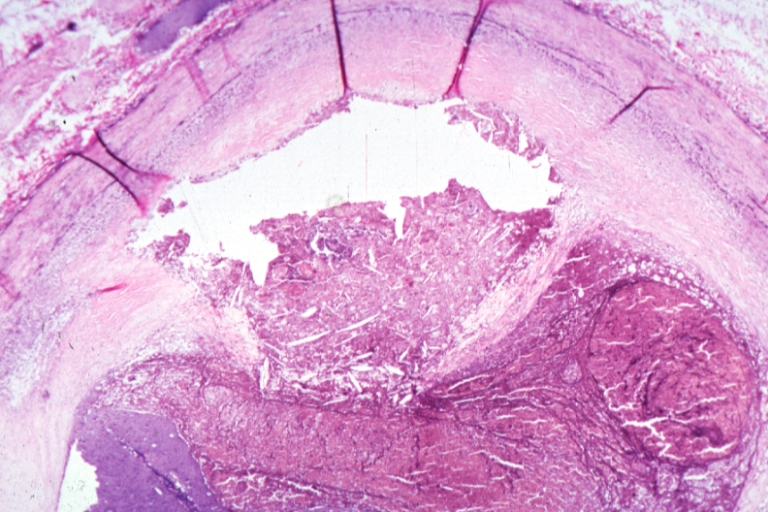
Coronary artery: Atherosclerosis: Micro, H&E, injected artery, marked large plaque with thin fibrous cap has eroded to adventitia, the start of an aneurysm
-
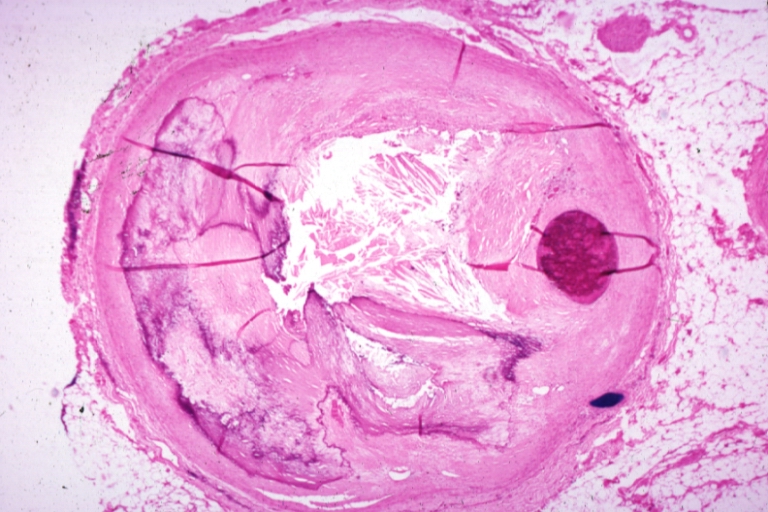
Coronary artery: Atherosclerosis: Micro, H&E, low mag, an excellent example of plaque with calcification and marked narrowing of lumen. 90% lumen is occluded by thrombus




-
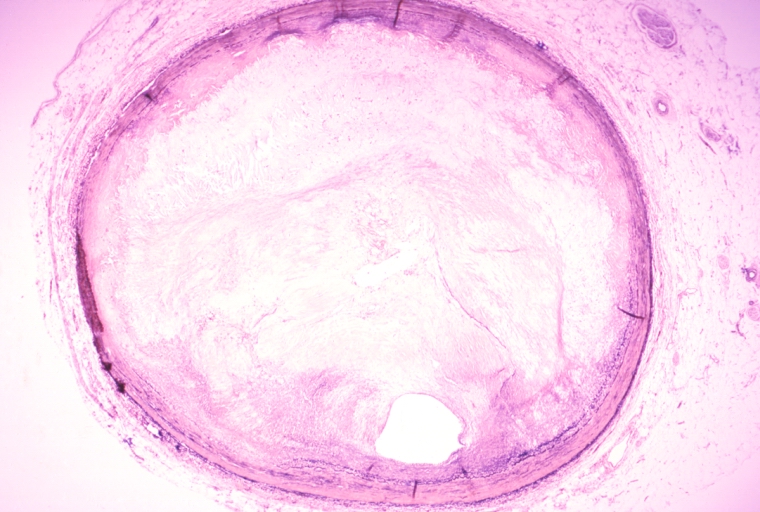
Coronary artery: Atherosclerosis: Micro, low mag, an excellent example of atheromatous plaque causing marked lumen obstruction. An uncomplicated plaque
-
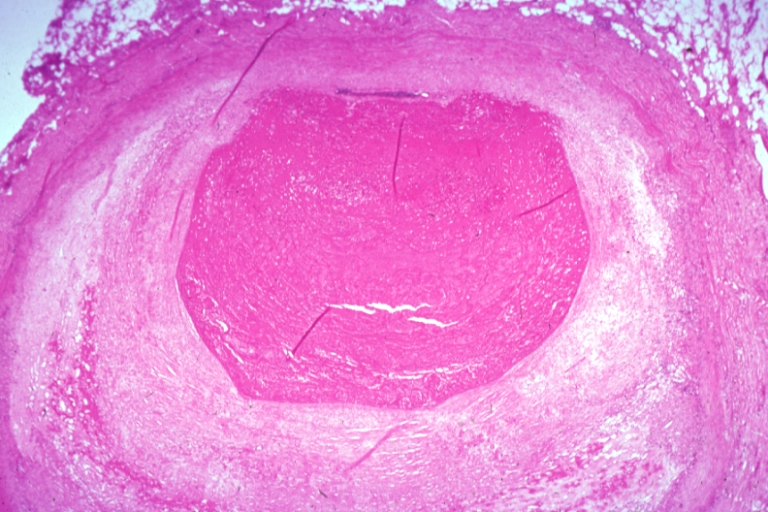
Coronary artery: Atherosclerosis: Micro, H&E, low mag, an excellent example of athero plaque. the lumen is completely occluded
-
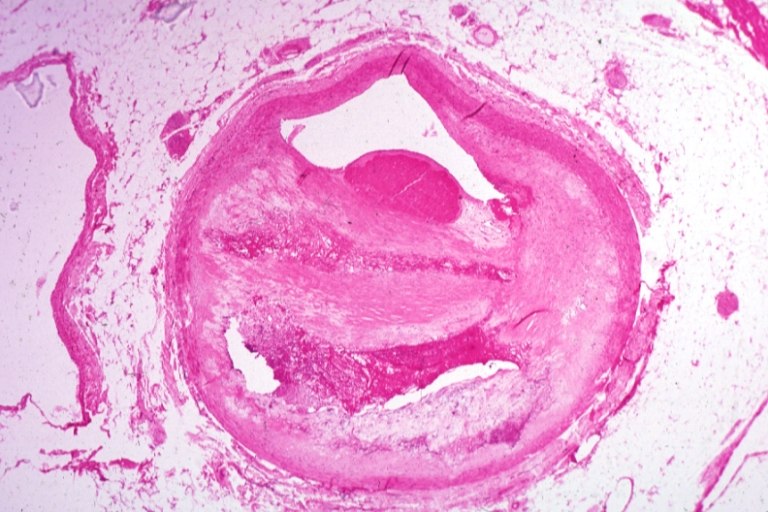
Coronary artery: Atherosclerosis: Micro, H&E, low mag, a plaque with hemorrhage and organizing mural thrombus in lumen
-
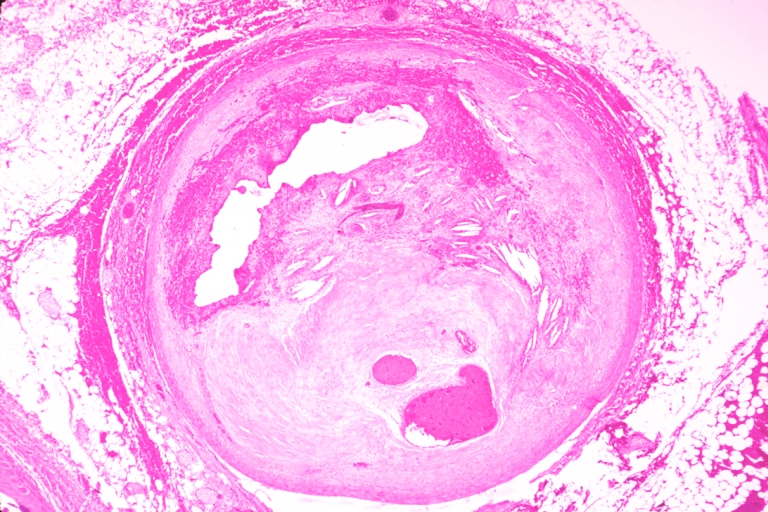
Coronary artery: Atherosclerosis: Micro, H&E, low mag, a good example of plaque with old hemorrhage and marked lumen compensation




-
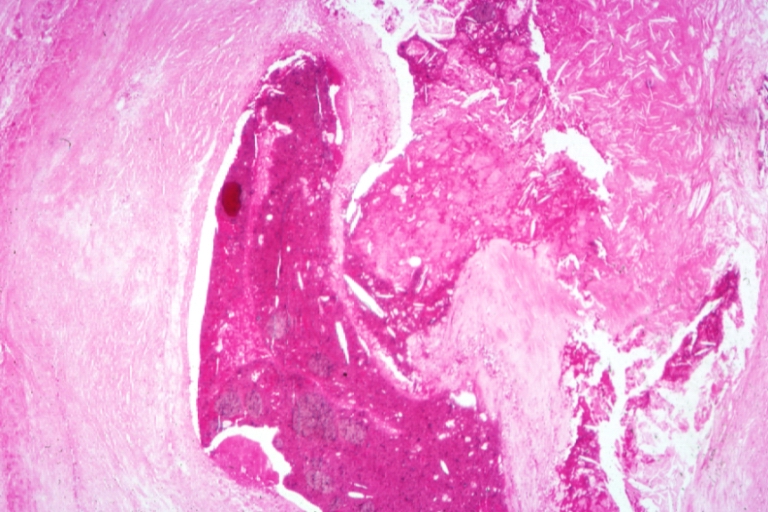
Coronary artery: Atherosclerosis: Micro, H&E, med mag, plaque rupture with thrombosis
-
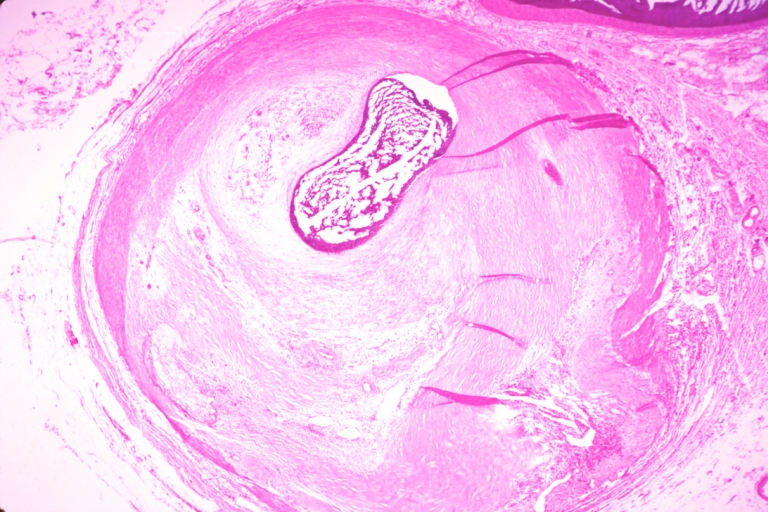
Coronary artery: Atherosclerosis: Micro, H&E, low mag, injected artery, a good example of atheromatous plaque that appears to owe much of its mass to an organized mural thrombus
-
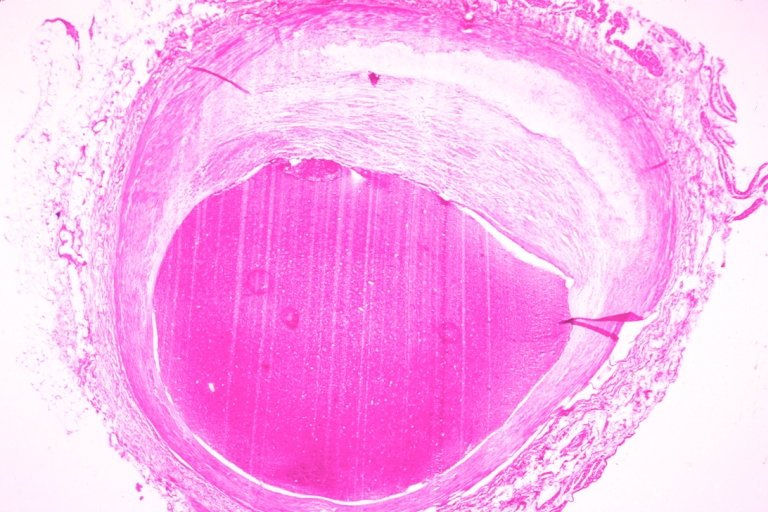
Coronary artery: Atherosclerosis: Micro, H&E, low mag, typical uncomplicated fibrous plaque
-
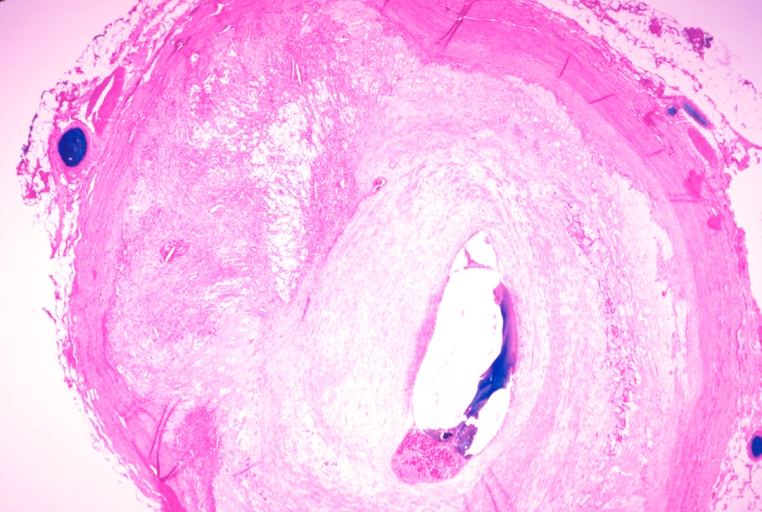
Coronary artery: Atherosclerosis: Micro, H&E, low mag, a good example of athero plaque with marked lumen narrowing. A small mural thrombus in lumen




-
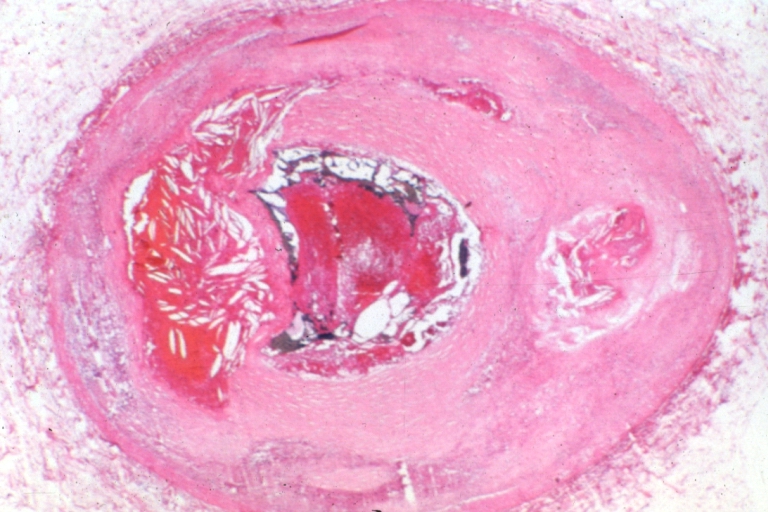
Coronary artery: Atherosclerosis: Micro, low mag, hemorrhage into plaque and thrombosis
-
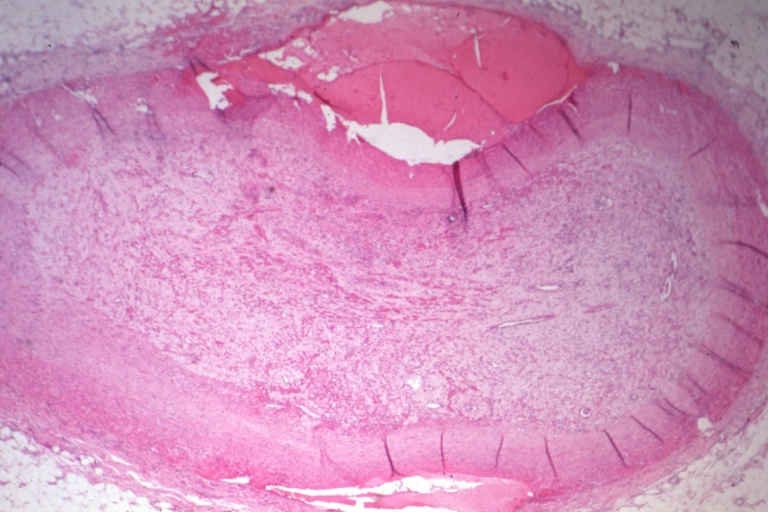
Coronary artery: Atherosclerosis: Recanalized Thrombus: Micro, low mag, H&E, lesion is nearly completely organized, small adventitial dissection probably related to bypass surgery about 24 hours before death (a good example)
-
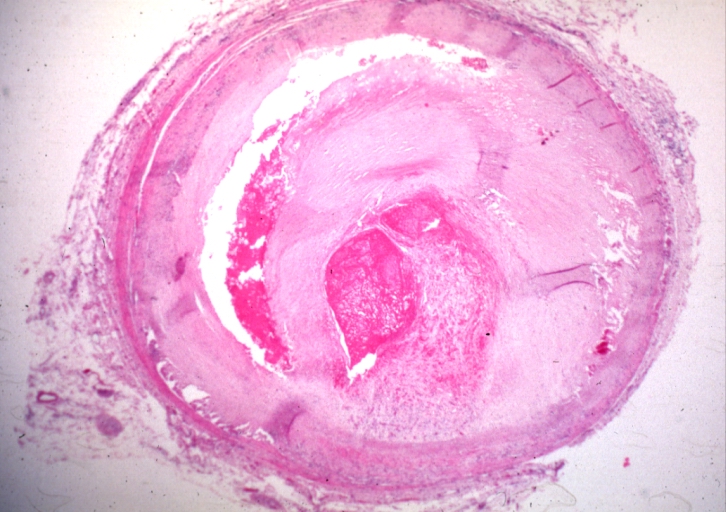
Coronary artery: Atherosclerosis: Plaque Hemorrhage and Thrombosis: Micro, low mag, H&E, quite good photo, large plaque with hemorrhage, organizing thrombus in lumen
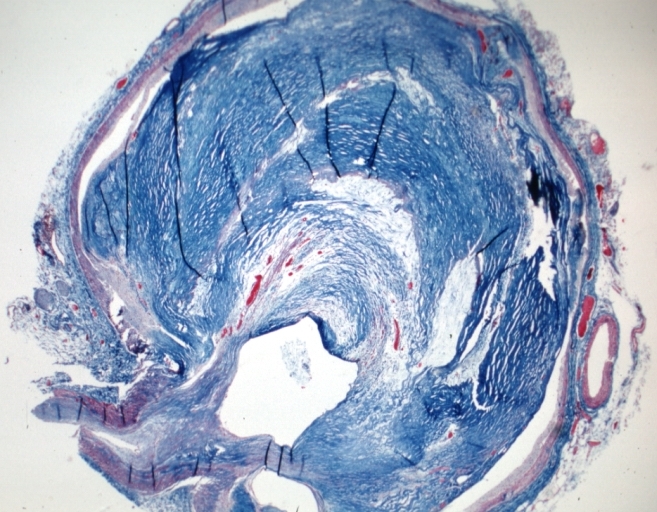
-
Coronary artery: Atherosclerosis, Severe: Micro, low mag, H&E, more than 90% stenosis, also shown a side branch




Aorta
-
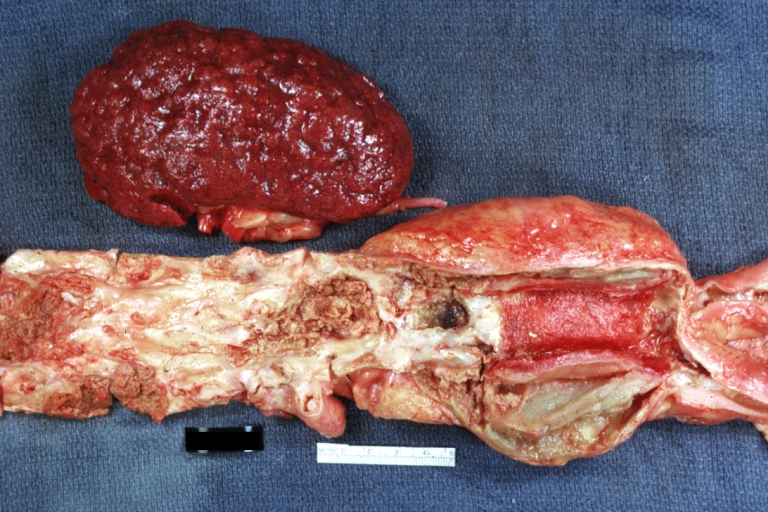
Kidney: Atheromatous Embolus: Gross, natural color, external view of kidney with typical scarring pattern of repeated infarction and aorta with severe atherosclerosis (quite good example)
-
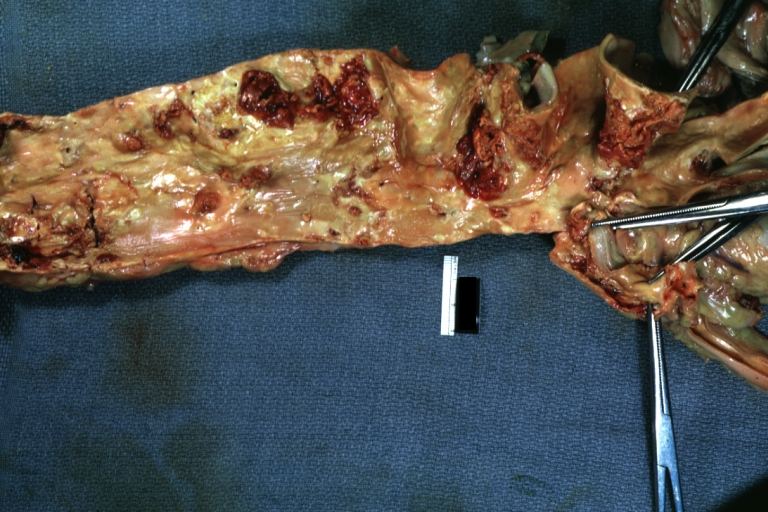
Aorta, Atherosclerosis: Gross, natural color, opened thoracic segment showing sessile plaques covering intima with several mural thrombi
-
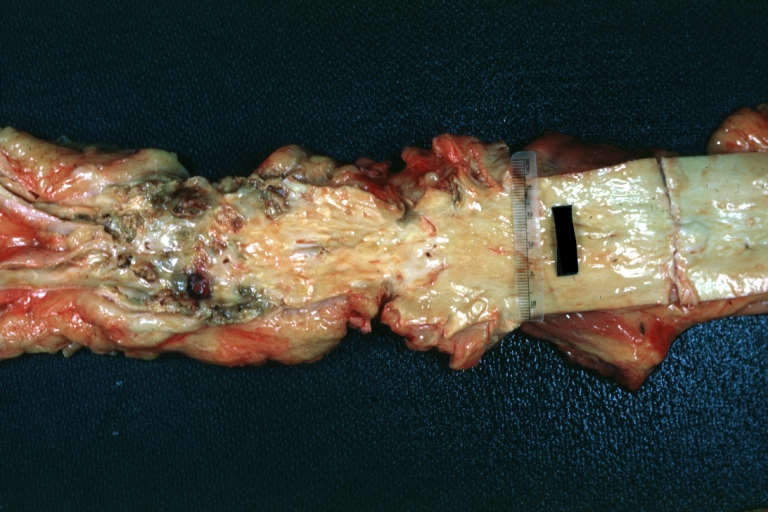
Aorta, Atherosclerosis: Gross natural color view of descending thoracic and abdominal segments with expected distribution of atherosclerotic lesions in subject over 60 years old. Not much in thoracic segment and extensive plaques in abdominal segment, some with ulceration
-
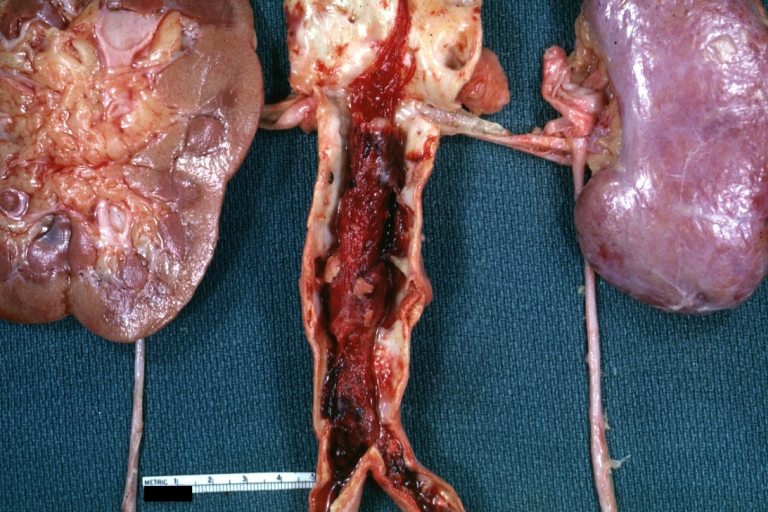
Thrombus: Gross natural color aorta with kidneys showing thrombotic occlusion due to atherosclerosis beginning just below renal arteries and extending into common iliac (very good example)




Carotid Artery
-
Carotid artery: Atherosclerosis: Gross, internal carotid plaque with thrombosis
-
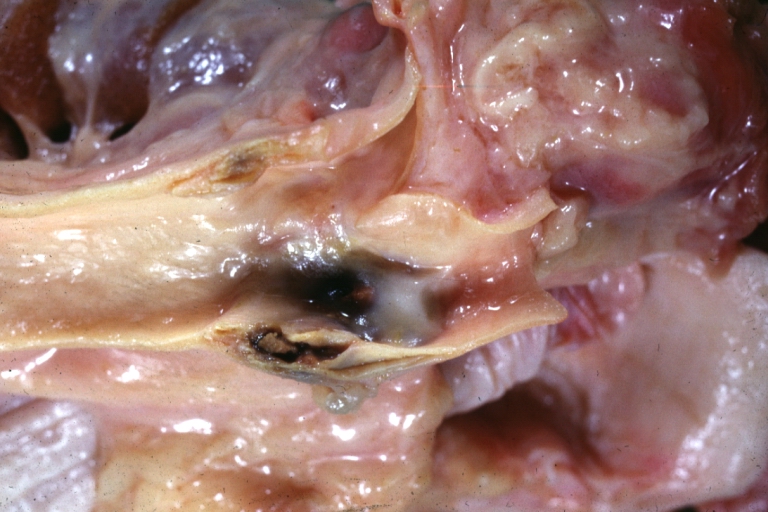
Carotid artery: Atherosclerosis: Gross, plaque with hemorrhage in carotid bulb
-
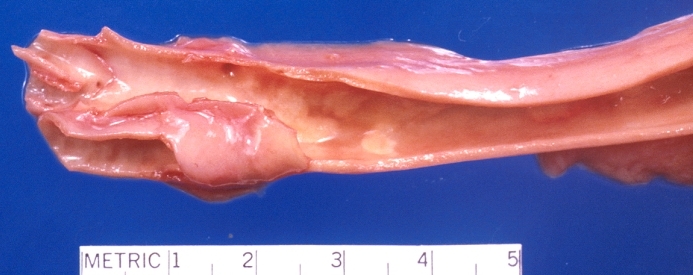
Carotid artery bifurcation, atherosclerosis
-
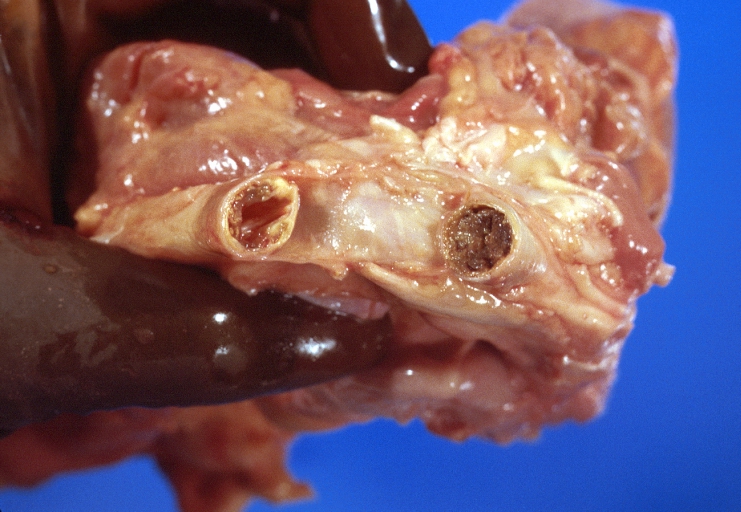
Carotid artery, atherosclerosis and thrombosis
-
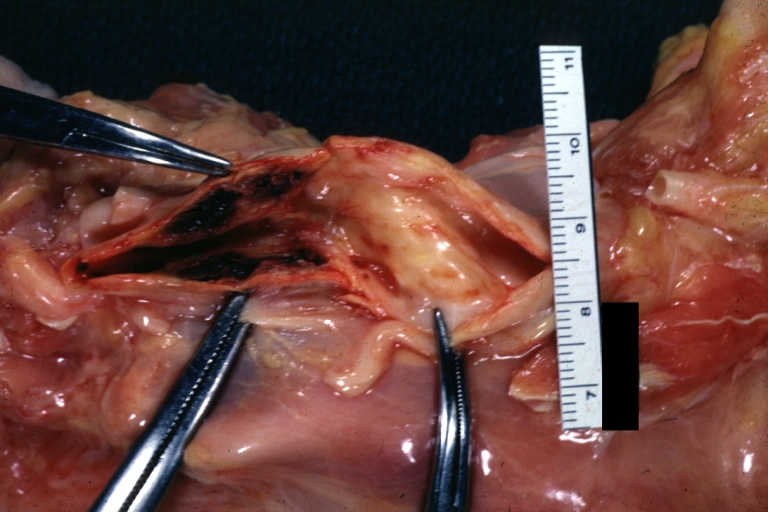
Carotid artery: Atherosclerosis: Gross, good example of carotid bulb plaque with thrombus




Lung
-
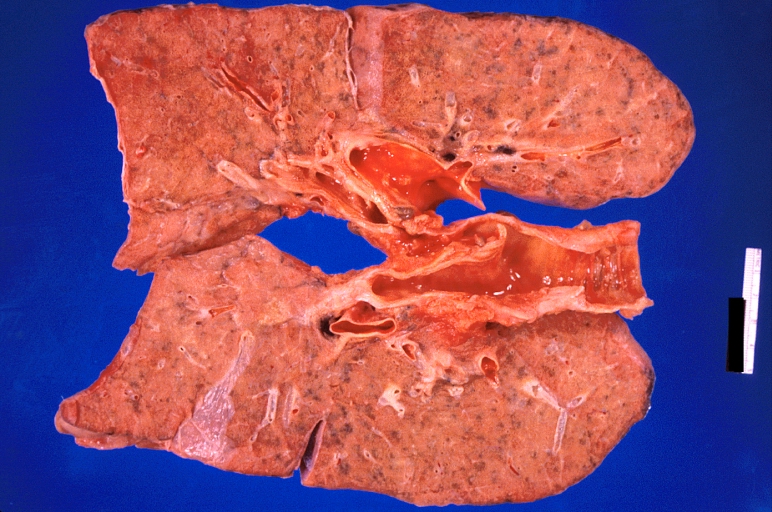
Lung: Pulmonary fibrosis and atherosclerosis of pulmonary artery
-
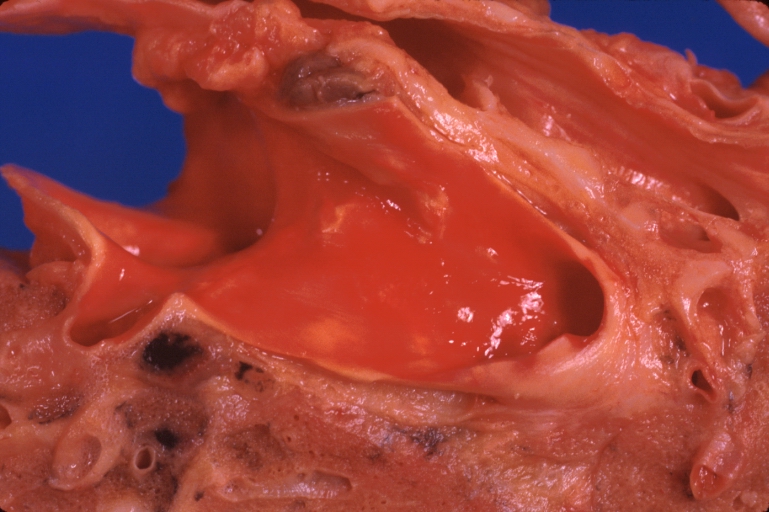
Lung: Pulmonary fibrosis and atherosclerosis of pulmonary artery
-
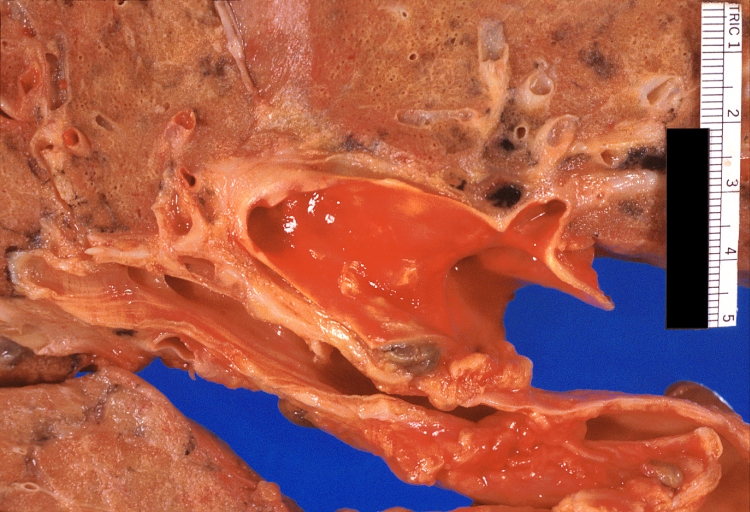
Lung: Pulmonary fibrosis and atherosclerosis of pulmonary artery
-
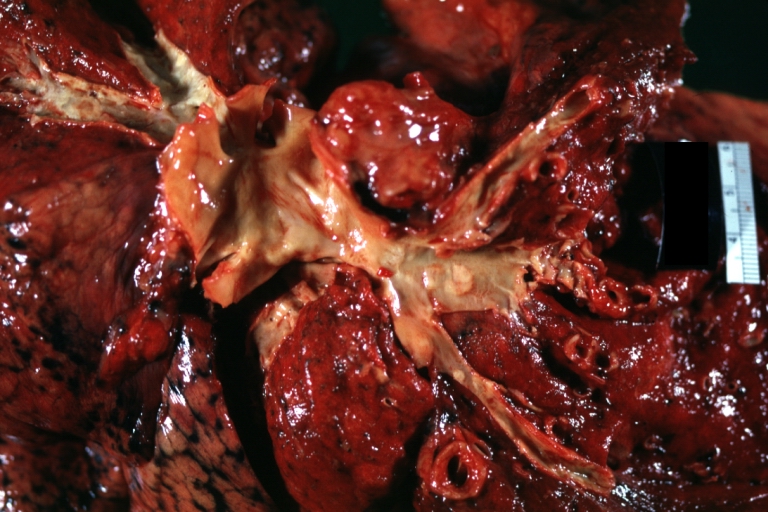
Lung: Atherosclerosis: Gross, natural color, arteries show atherosclerotic plaque lesions




Pulmonary Artery
-
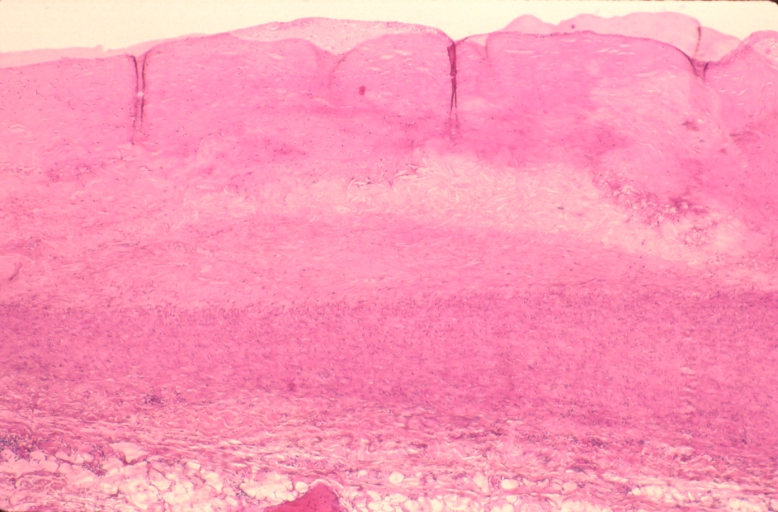
Pulmonary artery atherosclerosis in patient with pulmonary hypertension
-
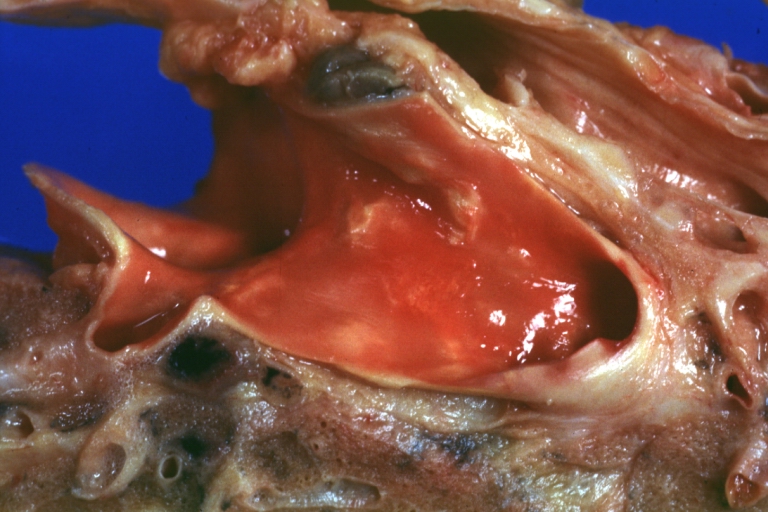
Pulmonary Artery Atherosclerosis: Gross, essentially natural color, artery stained by hemolysis, small plaque lesions
-
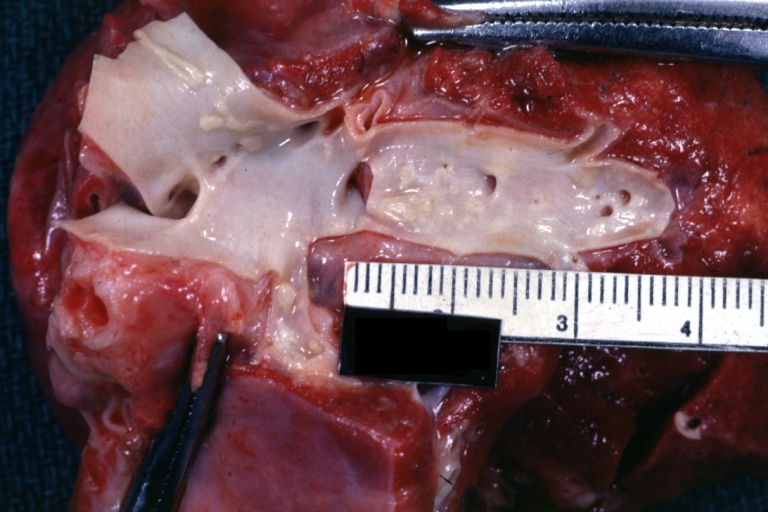
Atherosclerosis: Gross, natural color, close-up fatty plaques in large pulmonary artery
-
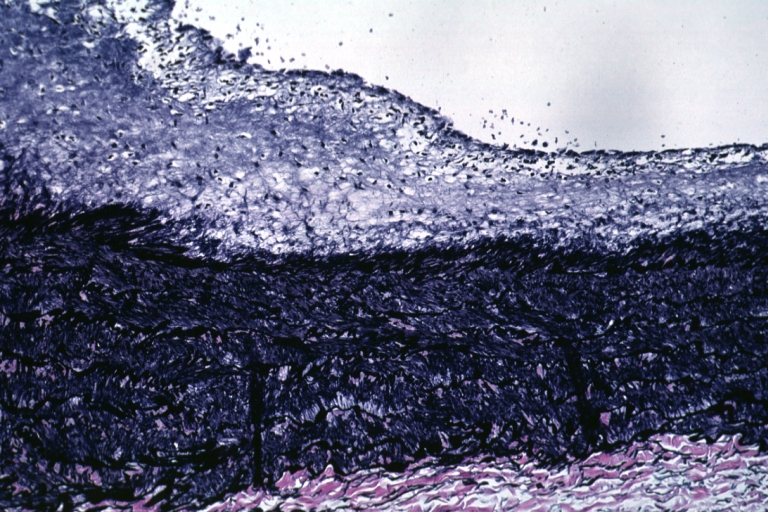
Atherosclerosis: Micro, low mag, van Gieson, thickened intima with fatty streak type lesion and preservation of fetal medial structure, a large pulmonary artery, 4yo male with primary pulmonary hypertension




Brain
-
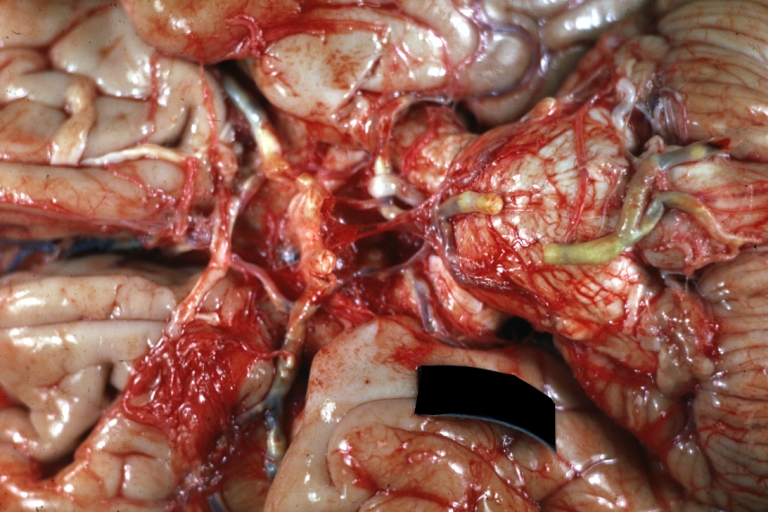
Brain: Atherosclerosis: Gross, a good example of atherosclerosis in vessels at base of brain.
-
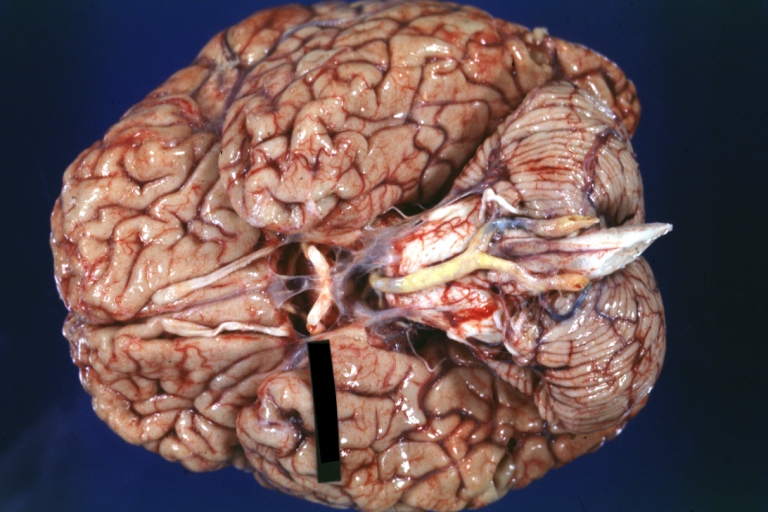
Brain: Basilar Artery Atherosclerosis: Gross, fixed tissue, an external view of base of brain (typical lesion)
-
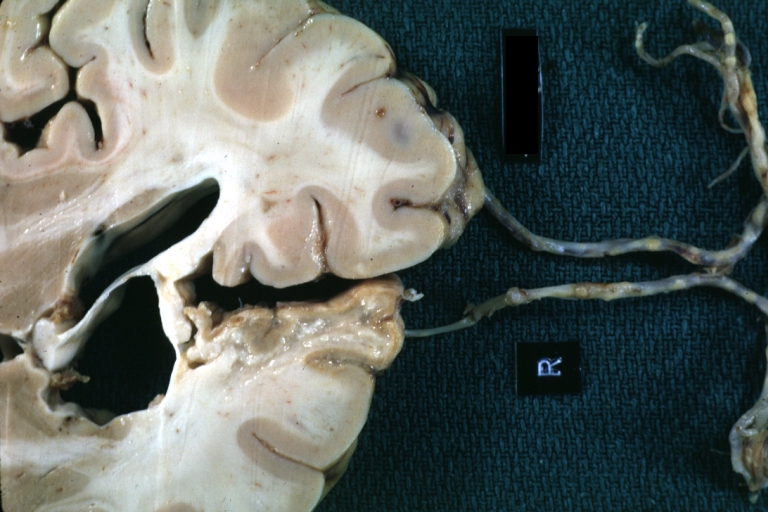
Brain: Old Cystic Encephalomalacia: Gross, fixed tissue, frontal lobe lesion with frontal artery atherosclerosis
-
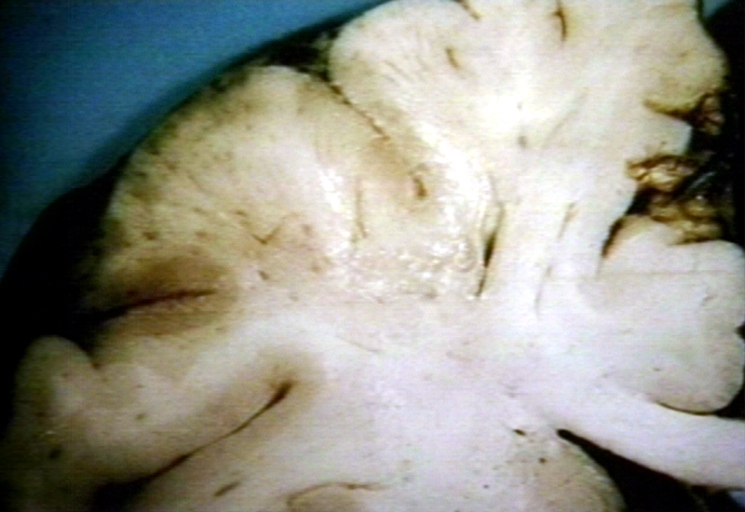
Brain: Watershed Infarct and Carotid Atherosclerosis




Kidney
-
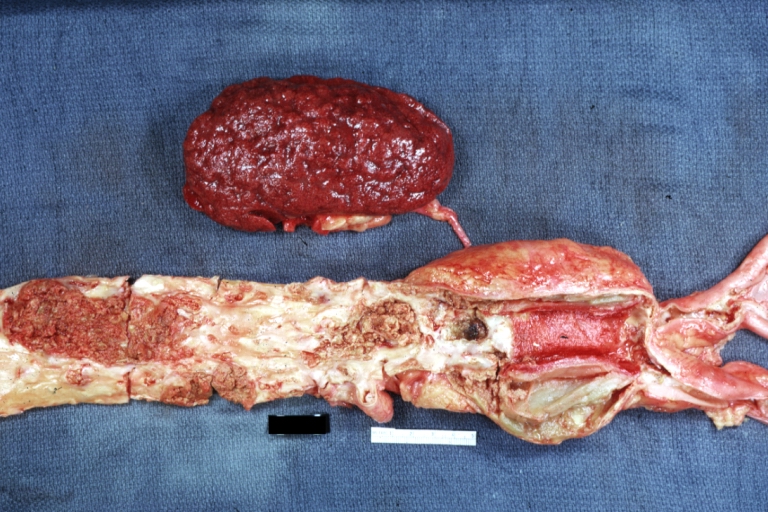
Atherosclerosis: Kidney: Atheromatous Embolus: Gross, natural color, kidney with typical scarring pattern of repeated embolism and aorta with severe atherosclerosis (a quite good example)
-
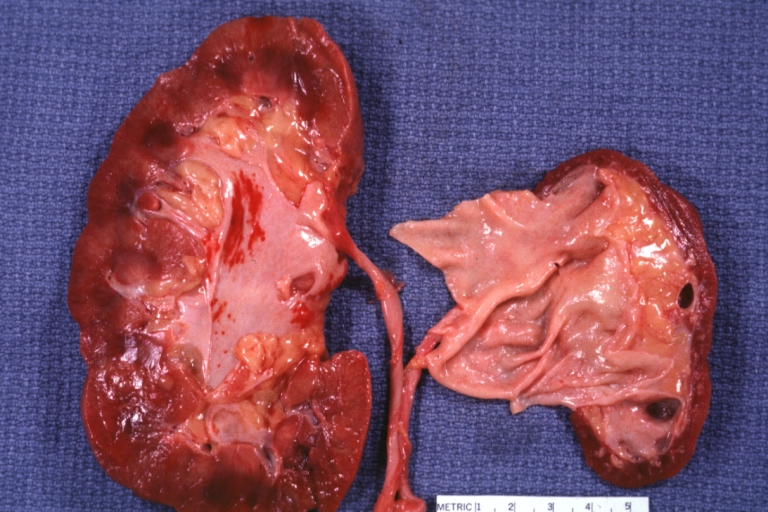
Atherosclerosis: Kidney: Atrophy secondary to renal artery atherosclerosis: Gross, natural color, both kidneys one very atrophic the large left kidney weighed 220 grams and the small left one 90 gram


References
- ↑ Weber, C.; Noels, H. (2011). "Atherosclerosis: current pathogenesis and therapeutic options". Nat Med. 17 (11): 1410–22. doi:10.1038/nm.2538. PMID 22064431.
- ↑ Davies, MJ.; Woolf, N.; Rowles, PM.; Pepper, J. (1988). "Morphology of the endothelium over atherosclerotic plaques in human coronary arteries". Br Heart J. 60 (6): 459–64. PMID 3066389. Unknown parameter
|month=ignored (help) - ↑ O'Brien, KD.; Olin, KL.; Alpers, CE.; Chiu, W.; Ferguson, M.; Hudkins, K.; Wight, TN.; Chait, A. (1998). "Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins". Circulation. 98 (6): 519–27. PMID 9714108. Unknown parameter
|month=ignored (help) - ↑ Kockx, MM.; De Meyer, GR.; Muhring, J.; Jacob, W.; Bult, H.; Herman, AG. (1998). "Apoptosis and related proteins in different stages of human atherosclerotic plaques". Circulation. 97 (23): 2307–15. PMID 9639374. Unknown parameter
|month=ignored (help) - ↑ Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 1987;316:131-1375. PMID