Amyloidosis: Difference between revisions
m (Robot: Changing Category:Disease state to Category:Disease) |
m (Robot: Changing Category:Diseases to Category:Disease) |
||
| Line 219: | Line 219: | ||
{{WikiDoc Sources}} | {{WikiDoc Sources}} | ||
[[Category: | [[Category:Disease]] | ||
[[Category:Rheumatology]] | [[Category:Rheumatology]] | ||
[[Category:Cardiology]] | [[Category:Cardiology]] | ||
[[Category:Mature chapter]] | [[Category:Mature chapter]] | ||
[[Category:Metabolic disorders]] | [[Category:Metabolic disorders]] | ||
[[Category:Inborn errors of metabolism]] | [[Category:Inborn errors of metabolism]] | ||
[[Category:Endocrinology]] | [[Category:Endocrinology]] | ||
Revision as of 18:48, 12 December 2011
For patient information, click here
| Amyloidosis | |
 | |
|---|---|
| Small bowel duodenum with amyloid deposition congo red 10X | |
| ICD-10 | E85 |
| ICD-9 | 277.3 |
| DiseasesDB | 633 |
| eMedicine | med/3377 med/3888 |
| MeSH | D000686 |
|
WikiDoc Resources for Amyloidosis |
|
Articles |
|---|
|
Most recent articles on Amyloidosis Most cited articles on Amyloidosis |
|
Media |
|
Powerpoint slides on Amyloidosis |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Amyloidosis at Clinical Trials.gov Clinical Trials on Amyloidosis at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Amyloidosis
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Amyloidosis Discussion groups on Amyloidosis Patient Handouts on Amyloidosis Directions to Hospitals Treating Amyloidosis Risk calculators and risk factors for Amyloidosis
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Amyloidosis |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
In medicine, amyloidosis refers to a variety of conditions in which amyloid proteins are abnormally deposited in organs and/or tissues, causing disease. A protein is amyloid if, due to an alteration in its secondary structure, it takes on a particular insoluble form, called the beta-pleated sheet.
Approximately 25 different proteins are known that can form amyloid in humans. Most of them are constituents of the plasma.
Different amyloidoses can be systemic (affecting many different organ systems) or organ-specific. Some are inherited, due to mutations in the precursor protein. Other, secondary forms are due to different diseases causing overabundant or abnormal protein production-such as with over production of immunoglobulin light chains in multiple myeloma (termed AL amyloid), or with continuous overproduction of acute phase proteins in chronic inflammation (which can lead to AA amyloid).
Definitions
What is Amyloid?
Amyloid is a pathological extracellular deposit composed predominantly of amyloid fibrils with characteristic morphology and pathognomonic tinctorial properties, specifically the capacity to bind Congo Red dye and then display red-green dichroism when viewed in strong cross polarised light. Amyloid fibrils are formed from normally soluble autologous precursor proteins that have undergone misfolding and aggregated into fibrils with a common cross Beta-sheet core structure.
The deposits are rich in proteoglycans and always contain the non-fibrillar normal plasma glycoprotein, serum amyloid P component (SAP), which binds specifically to all amyloid fibrils.
What is Amyloidosis?
Amyloidosis is disease caused by amyloid deposits in the tissues. The deposits may be local or systemic in distribution and acquired or hereditary in aetiology. All patients with amyloidosis have amyloid deposits in their tissues when they first present clinically. In each individual, disease is always worse when there is more amyloid present, and arrest or regression of amyloid deposition is associated with clinical benefit. Only organs or tissues in which amyloid deposits are present are directly functionally compromised, even when the amyloid fibril precursor protein is ubiquitous.
What is not Amyloidosis?
Several important diseases are associated with amyloid deposits but are not amyloidosis.
The neuropathology of Alzheimer’s disease by definition includes intra-cerebral ABeta amyloid deposits, but it is not known whether these deposits are responsible for the neuronal cell death that causes cognitive decline; non-fibrillar ABeta aggregates may be more directly involved. In contrast, by analogy with the notable friability of systemic blood vessels containing AL amyloid deposits, it seems likely that the cerebrovascular ABeta amyloid deposits in cerebral amyloid angiopathy are responsible for cerebral haemorrhage.
Most patients with type 2 diabetes have amyloid deposits in the islets of Langerhans, which may exacerbate islet dysfunction, but amyloid is not the original cause of their diabetes. Cerebral amyloid deposits of the prion protein are present in many forms of transmissible spongiform encephalopathy but are absent in others, including bovine spongiform encephalopathy in cows and fatal familial insomnia in humans, and amyloid is thus not essential for pathogenesis of TSE.
Many other diseases are associated with, and possibly caused by, protein misfolding and aggregation. Some of these aggregates share the amyloid cross Beta-sheet fold, for example in Huntington’s and related poly-glutamine repeat hereditary neurodegenerative diseases, and in Parkinson’s disease.
However intracellular and intranuclear protein aggregates have very different pathogenetic effects than extracellular amyloid deposits, and are in radically different locations for drug intervention. They are also not associated with the non-fibrillar components of amyloid deposits, the proteoglycans and SAP. These intracellular protein aggregates are thus not amyloid and the diseases with which they are associated are not amyloidosis. Although there may be informative similarities and parallels, especially in protein misfolding and aggregation studied in vitro, to conflate such widely different processes is misleading and potentially dangerous when extrapolated to development of therapeutic interventions.
Other protein misfolding diseases, such as the serpinopathies, are even more remote from amyloidosis, with intracellular deposition of protein aggregates that do not share the typical amyloid cross-Beta fold and where much of the pathogenesis of disease is related to loss of normal serpin function rather than adverse effects of the aggregates.
What is SAP?
SAP is a highly conserved constitutive normal trace plasma protein with specific avid calcium dependent binding to all amyloid fibrils that causes its remarkable selective concentration and persistence in amyloid deposits of all types.
SAP is intrinsically resistant to proteolysis and is further stabilised by its tight binding to amyloid fibrils, which in turn stabilises the fibrils and protects them from proteolysis.
SAP knockout mice show delayed and reduced deposition of experimentally induced reactive systemic amyloid. SAP is therefore a valid therapeutic target, and in vivo inhibition of SAP binding and/or depletion of circulating SAP should reduce amyloid burden.
Pathophysiology
In amyloidosis the link between physical presence of amyloid deposits and disease is irrefutably strong. Pre-fibrillar protein aggregates are cytotoxic for cultured cells exposed under artefactual in vitro conditions but there is no evidence that this is pathophysiologically relevant to amyloidosis in vivo. No clinical or pathological effects are detectable in any form of acquired or hereditary systemic amyloidosis until amyloid deposits are demonstrable, regardless of the length of time that the predisposition exists before amyloid develops.
Clinical impairment progresses as amyloid deposits increase in size, and regression of amyloid is associated with clinical improvement and survival. If damaged organs are replaced there is no organ dysfunction, even in the face of unchanged abundance of amyloid fibril precursor proteins fully capable of aggregation and fibril formation, until new amyloid deposits form and accumulate to a critical mass. This usually takes years.
There is usually no histological evidence of inflammation, or cell death or dysfunction, even in massively amyloid laden organs, and the organs continue to function until the amyloid deposits critically compromise structure and/or function. For example, cardiac amyloid deposition impairs diastolic filling by stiffening the ventricle.
There is no evidence of cytotoxicity in the myocardium and the heart muscle continues to contract excellently and propel the progressively reduced stroke volume until the end. Similarly in hereditary transthyretin amyloidosis, amyloid fibrils may accumulate over years in the vitreous humour of the eye until fibril density impairs optical clarity and causes blindness. The amyloidogenic variant transthyretin in the eye is synthesised in the choroid plexus and by the ciliary epithelium directly overlying the retina, and continuously bathes the retina. However replacement of the amyloid laden vitreous with optically clear medium restores perfect vision in every case! The putative toxic pre-fibrillar aggregates evidently have no deleterious effect on retinal neurones, whereas accumulation of amyloid fibrils causes blindness and their removal cures it.
Finally there are many clinical situations in which the local physical presence of substantial amyloid deposits alone is the only possible cause of disease and death, and where reduction or removal of the deposits relieves symptoms and signs and is life saving. [1]
Classification of amyloid
There are 15 biologically distinct forms of amyloid, some more clinically significant than others. Following is a brief description of the more common types of amyloid:
| Amyloid type | Description |
|---|---|
| AL, amyloid light chain | Contains immunoglobulin light-chains (λ,κ) derived from plasma cells |
| AA, amyloid associated | Non-immunoglobulin protein made in the liver |
| Aβ | Found in Alzheimer disease brain lesions |
| TTR, Transthyretin | A mutant form of a normal serum protein that is deposited in the genetically determined familial amyloid polyneuropathies. TTR is also deposited in the heart in senile systemic amyloidosis. |
| β2 microglobulin | Not to be confused with Aβ, β2 is a normal serum protein, part of major histocompatability complex (MHC) Class 1 molecules. Can occur in long term haemodialysis. |
| PrP, Prion related protein | In Prion diseases, misfolded prion proteins deposit in tissues and resemble amyloid proteins. |
Diagnosis
Amyloid can be diagnosed on histological examination of affected tissue. Amyloid deposits can be identified histologically by Congo red staining and viewing under polarized light where amyloid deposits produce a distinctive 'apple green birefringence'. Further, specific, tests are available to more precisely identify the amyloid protein. Biopsies are taken from affected organs (for example, the kidney), or often in the case of systemic amyloid, from the rectum or anterior abdominal adipose tissue.
In addition, all amyloid deposits contain serum amyloid P component (SAP), a circulating protein of the pentraxin family. Radionuclide SAP scans have been developed which can anatomically localize amyloid deposits in patients.
Systemic amyloidosis
Primary/Hereditary amyloidosis
These rare hereditary disorders are usually due to point mutations in precursor proteins, and are also usually autosomal dominantly transmitted.The precursor proteins are;
- transthyretin-the most commonly implicated protein.
- lysozyme
- apolipoprotein B
- fibrinogen
- apolipoprotein A1
- gelsolin
Secondary amyloidosis
These are far more common than the primary amyloidoses.
- AL amyloidosis (immunoglobulin light chains are the precursor protein, overproduced in multiple myeloma). This is sometimes, confusingly and erroneously, called 'primary amyloidosis'.
- AA amyloidosis (the precursor protein is serum amyloid A protein (SAA), an acute-phase protein due to chronic inflammation). In contrast to AL amyloid, this has previously been termed 'secondary amyloidosis'.These occur with a wide variety of diseases associated with chronic inflammation, such as Rheumatoid arthritis, Familial Mediterranean fever or chronic infection.
- Dialysis related amyloidosis (the precursor protein is beta-2-microglobulin which is not removed with dialysis, and thus accumulates in patients with end stage renal failure on dialysis).
Organ-specific amyloidosis
In almost all of the organ-specific pathologies, there is significant debate as to whether the amyloid plaques are the causal agent of the disease or instead a downstream consequence of a common idiopathic agent. The associated proteins are indicated in parentheses.
Neurological amyloid
- Alzheimer's disease (Aβ 39-43)
- Parkinson's disease (alpha-synuclein)
- Huntington's disease (huntingtin)
- Transmissible spongiform encephalopathies caused by prion protein (PrP) were sometimes classed as amyloidoses, as one of the four pathological features in diseased tissue is the presence of amyloid plaques. These diseases include;
- Creutzfeldt-Jakob disease (PrP in cerebrum)
- Kuru (diffuse PrP deposits in brain)
- Fatal Familial Insomnia (PrP in thalamus)
- Bovine spongiform encephalopathy (PrP in cerebrum of cows)
Cardiovascular amyloid
- Cardiac amyloidosis
- See the chapter on CMR in Cardiac Amyloidosis
- Senile cardiac amyloidosis-may cause heart failure
- Congophilic angiopathy
Other
- Amylin deposition can occur in the pancreas in some cases of type 2 diabetes mellitus
Differential Diagnosis of Other Conditions to be Distinguished from Amyloidosis
- Acute myocarditis
- Bechterew's Disease
- Bronchiectasis
- Carpal Tunnel Syndrome
- Collagen Vascular Disease
- Drug/toxic nephropathy
- Familial Mediterranean Fever
- Glomerulonephritis
- Hemodialysis Amyloidosis
- Interstitial lung diseases
- Leprosy
- Monoclonal gammopathies
- Multiple Myeloma
- Myocardial fibrosis
- Nephrotic Syndrome
- Osteomyelitis
- Peripheral neuropathy
- Restrictive cardiomyopathy
- Rheumatoid Arthritis
- Rheumatoid Polyarteritis
- Syphilis
- Systemic Lupus Erythematosus
- Tuberculosis
- Ulcerative colitis
- Vitamin deficiencies
Treatment
Our goal in treatment of systemic amyloidosis: Clinical assessment and SAP scintigraphy in thousands of patients with systemic amyloidosis over the past 20 years, have established that sufficient reduction of the abundance of amyloid fibril precursor proteins, in both acquired and hereditary amyloidosis, is associated with arrest of amyloid deposition, regression of deposits, and clinical benefit.
Further measures to reduce amyloid deposition and/or promote regression will provide incremental benefits, hence our longstanding interest in SAP. [2] [3]
Examples
Images shown below are courtesy of Professor Peter Anderson and published with permission. © PEIR, University of Alabama at Birmingham, Department of Pathology
-
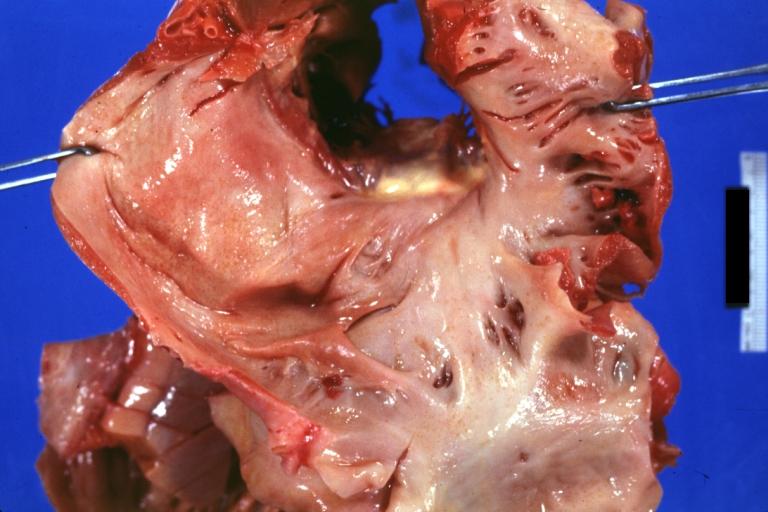
Amyloidosis Lesion In Left Atrium: Gross natural color view of a diagnostic lesion
-
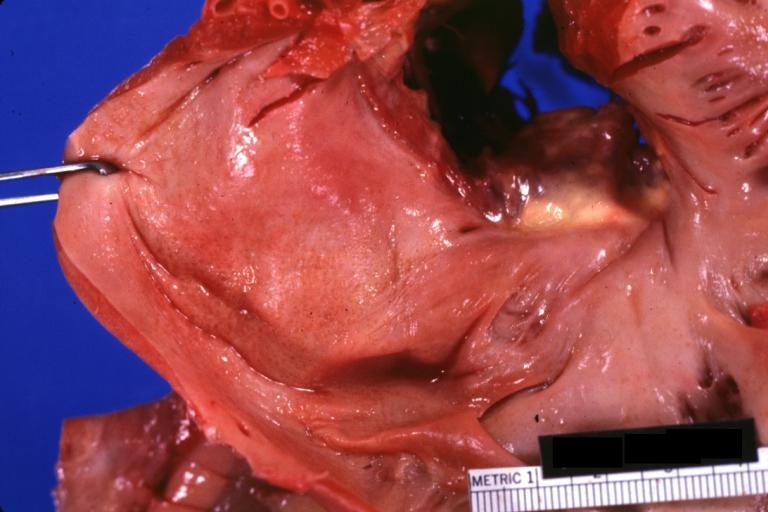
Amyloidosis Lesion In Left Atrium: Gross natural color close-up
-
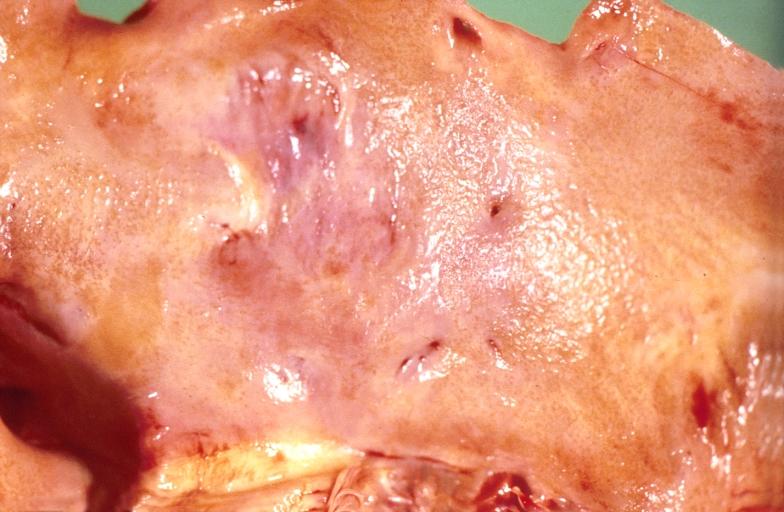
Amyloidosis, left atrium, endocardial nodules
-
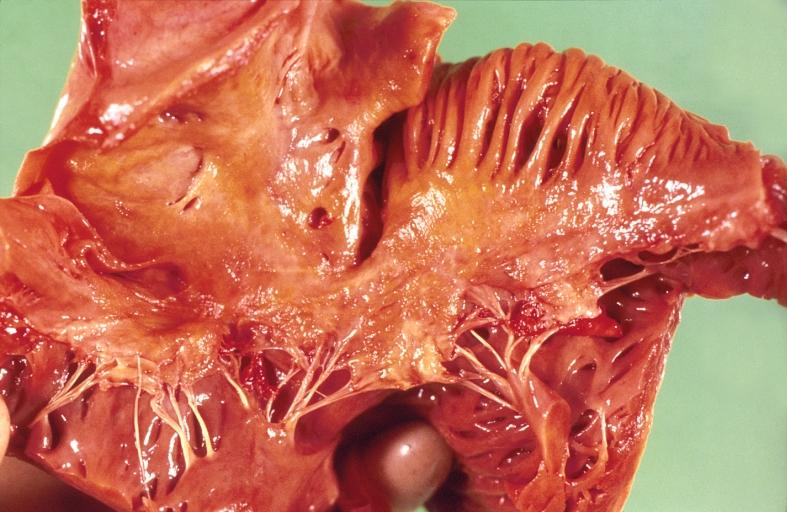
Amyloidosis, left atrium, endocardial nodules
-

Amyloidosis and left ventricular hypertrophy
-
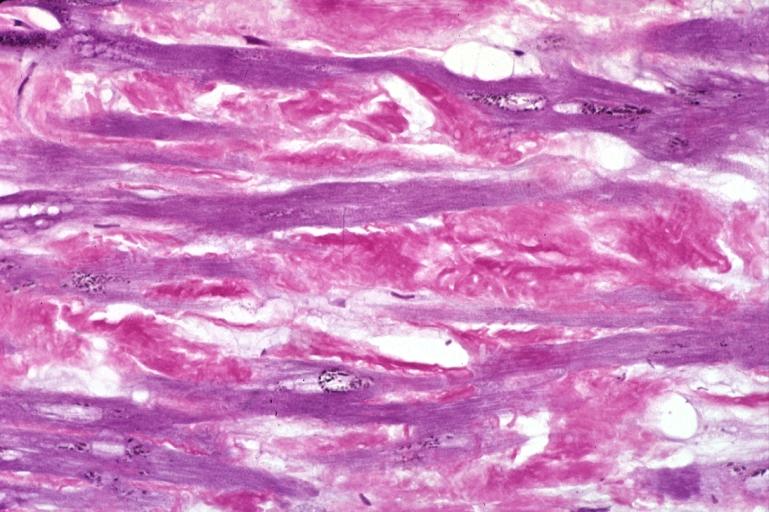
Heart: Amyloidosis, aldehyde fuchsin stain.
-
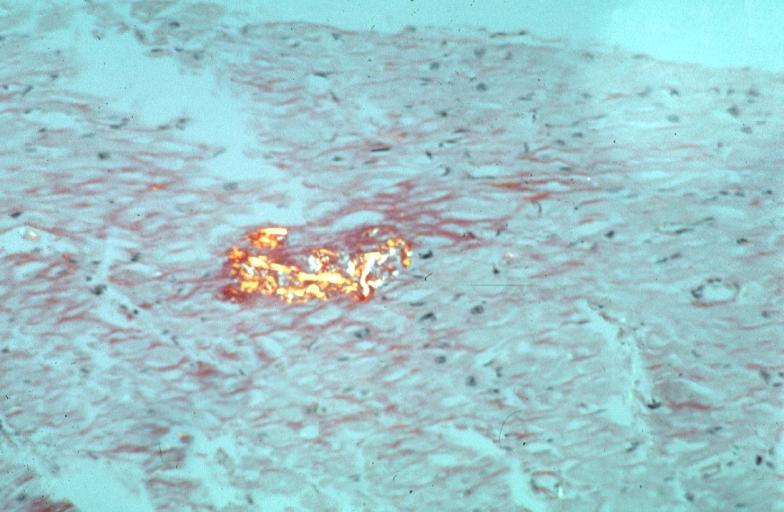
Heart: Perivascular amyloid, amyloidosis, congo red showing birefringence
-
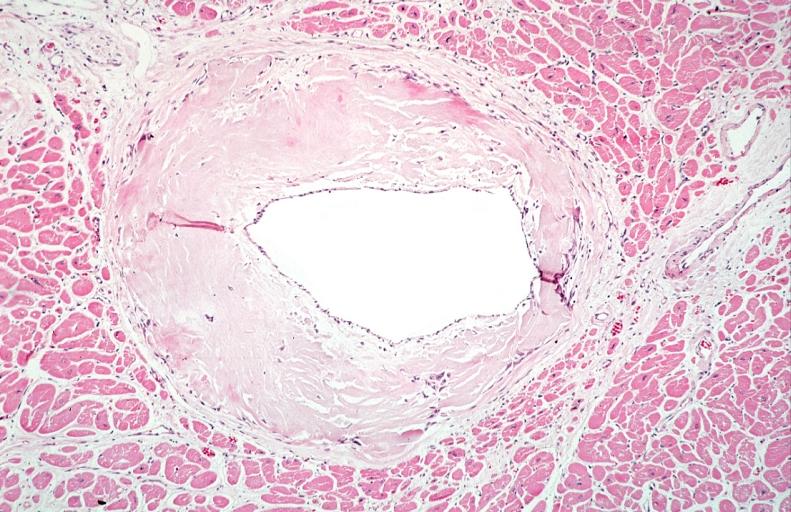
Heart: Perivascular amyloid, amyloidosis (Hematoxylin and eosin staining)
-

URINARY SYSTEM: Kidney, amyloidosis
-
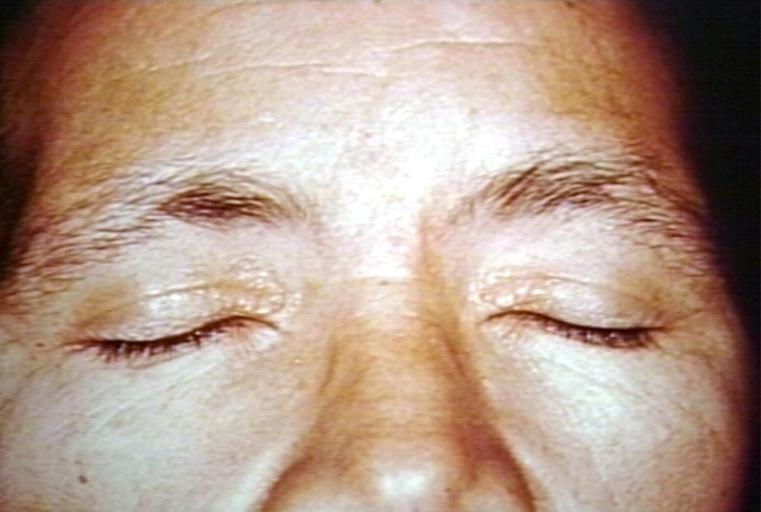
SKIN: AMYLOIDOSIS, PRIMARY; YELLOW LESIONS
-

TONGUE: PRIMARY AMYLOIDOSIS
-
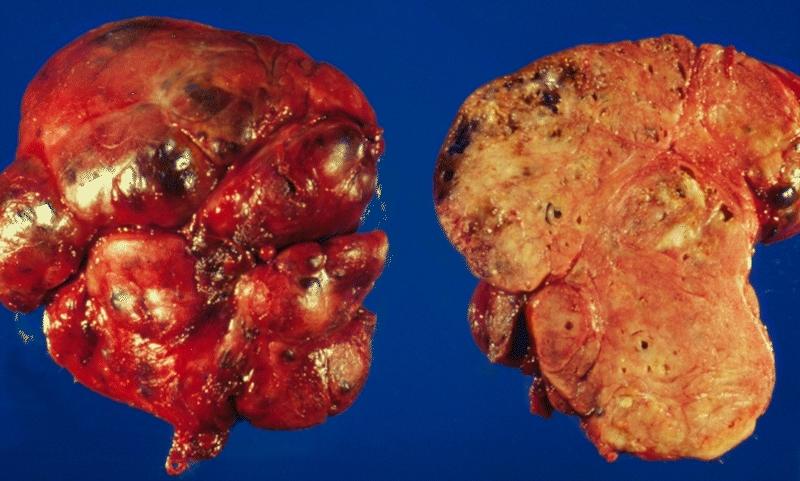
THYROID GLAND: AMYLOIDOSIS OF THE THYROID This illustration shows the outer aspect and cut surface of localized amyloidosis of the thyroid gland (amyloid tumor). The gland is enlarged and bosselated and exhibits a salmon color on cross section.
-
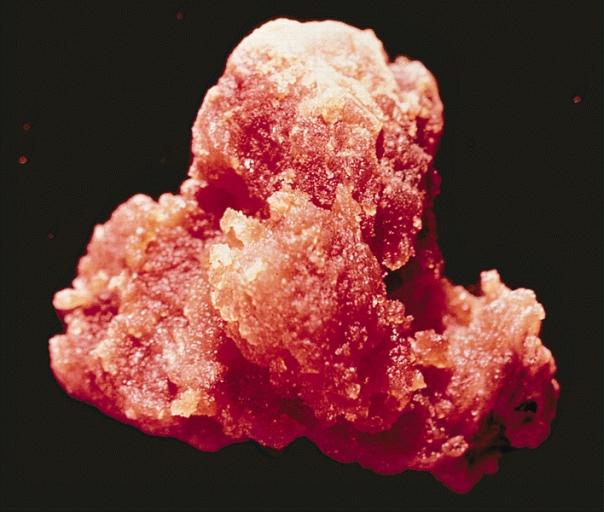
LOWER RESPIRATORY TRACT: NODULAR PARENCHYMAL AMYLOIDOSIS This nodule of amyloid shells taken out from the lung as an irregular, tan, waxy mass.
-

LIVER: Amyloidosis
-
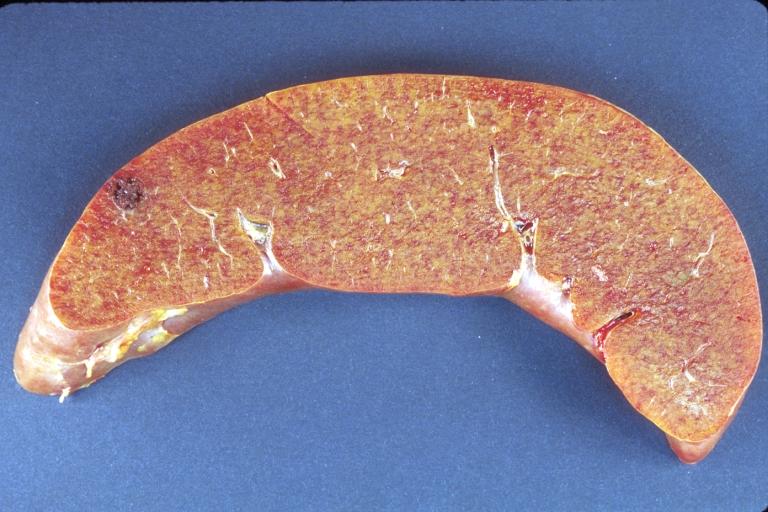
LIVER-BILIARY TRACT: Liver, amyloidosis
-

KIDNEY-BLADDER-URINARY: AMYLOIDOSIS OF URINARY BLADDER (Courtesy of Dr. George M. Farrow, Rochester, MN.)
















References
- ↑ Amyloid and amyloidosis. Gilles Grateau, Robert A. Kyle, Martha Skinner, 2005 ISBN 0-8493-3534-5
- ↑ Pepys, M.B. (1999) The Lumleian Lecture. C-reactive protein and amyloidosis: from proteins to drugs? In: Horizons in Medicine, Vol. 10 (Williams, G., eds.), Royal College of Physicians of London, London, pp. 397-414.
- ↑ Pepys, M.B., et al. (2002) Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature, 417: 254-259.
Template:Metabolic pathology Template:SIB de:Amyloidose et:Amüloidoos fi:Amyloidoosi