Parkinson's disease pathophysiology: Difference between revisions
(Created page with "__NOTOC__ {{Parkinson's disease}} {{CMG}} ==Overview== ==Pathophysiology== In the brain the direct pathway facilitates movement and the indirect pathway inhibits movement, ...") |
No edit summary |
||
| Line 22: | Line 22: | ||
The symptoms of Parkinson's disease result from the loss of pigmented [[dopamine]]-secreting (dopaminergic) cells, secreted by the same cells, in the [[substantia nigra|pars compacta]] region of the [[substantia nigra]] (literally "black substance"). These neurons project to the [[striatum]] and their loss leads to alterations in the activity of the neural circuits within the basal ganglia that regulate movement, in essence an inhibition of the [[direct pathway]] and excitation of the [[indirect pathway]]. | The symptoms of Parkinson's disease result from the loss of pigmented [[dopamine]]-secreting (dopaminergic) cells, secreted by the same cells, in the [[substantia nigra|pars compacta]] region of the [[substantia nigra]] (literally "black substance"). These neurons project to the [[striatum]] and their loss leads to alterations in the activity of the neural circuits within the basal ganglia that regulate movement, in essence an inhibition of the [[direct pathway]] and excitation of the [[indirect pathway]]. | ||
===Genetic=== | |||
In recent years, a number of specific genetic mutations causing Parkinson's disease have been discovered, including in certain populations ([[Contursi]], Italy). These account for a small minority of cases of Parkinson's disease. Somebody who has Parkinson's disease is more likely to have relatives that also have Parkinson's disease. However, this does not mean that the disorder has been passed on genetically. | |||
Genetic forms that have been identified include: | |||
:''external links in this section are to [[OMIM]]'' | |||
{| class="wikitable" | |||
| '''Type''' || '''OMIM''' || '''[[Locus (genetics)|Locus]]''' || '''Details''' | |||
|- | |||
| ''PARK1'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=168601 OMIM #168601] || 4q21 || caused by mutations in the ''[[SNCA]]'' gene, which codes for the [[protein]] [[alpha-synuclein]]. PARK1 causes [[autosomal dominant]] Parkinson disease. So-called ''PARK4'' ([http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=605543 OMIM #605543]) is probably caused by triplication of ''SNCA''.<ref>{{cite journal |author=Singleton AB, Farrer M, Johnson J, ''et al'' |title=alpha-Synuclein locus triplication causes Parkinson's disease |journal=Science |volume=302 |issue=5646 |pages=841 |year=2003 |pmid=14593171 |doi=10.1126/science.1090278}}</ref> | |||
|- | |||
| ''PARK2'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=602544 OMIM *602544] || 6q25.2-q27 || caused by mutations in protein [[Parkin (ligase)|parkin]]. Parkin mutations may be one of the most common known genetic causes of early-onset Parkinson disease. In one study, of patients with onset of Parkinson disease prior to age 40 (10% of all PD patients), 18% had parkin mutations, with 5% [[homozygous]] mutations.<ref>{{cite journal | author=Poorkaj P ''et al.'' | title=''parkin'' mutation analysis in clinic patients with early-onset Parkinson's disease | journal=American Journal of Medical Genetics Part A | year=2004 | volume=129A | | |||
issue=1 | pages= 44–50 | url=http://www3.interscience.wiley.com/cgi-bin/abstract/109062750/ABSTRACT?CRETRY=1&SRETRY=0}}</ref> Patients with an [[autosomal recessive]] family history of parkinsonism are much more likely to carry parkin mutations if age at onset is less than 20 (80% vs. 28% with onset over age 40).<ref>{{cite journal | author=Ebba Lohmann ''et al.'' | title=How much phenotypic variation can be attributed to parkin genotype? | journal=Annals of Neurology | year=2003 | volume=54 | issue=2 | pages= 176–185|url=http://www3.interscience.wiley.com/cgi-bin/abstract/104536414/ABSTRACT | pmid = 12891670}}</ref>Patients with [[parkin]] mutations (PARK2) do not have Lewy bodies. Such patients develop a syndrome that closely resembles the sporadic form of PD; however, they tend to develop symptoms at a much younger age. | |||
|- | |||
| ''PARK3'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=602404 OMIM %602404] || 2p13 || autosomal dominant, only described in a few kindreds. | |||
|- | |||
| ''PARK5'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=191342 OMIM +191342] || 4p14 || caused by mutations in the ''UCHL1'' gene which codes for the protein [[ubiquitin carboxy-terminal hydrolase L1]] | |||
|- | |||
| ''PARK6'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=605909 OMIM #605909] || 1p36 || caused by mutations in ''PINK1'' ([http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=605909 OMIM *608309]) which codes for the protein [[PTEN-induced putative kinase 1]]. | |||
|- | |||
| ''[[PARK7]]'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=606324 OMIM #606324] || 1p36 || caused by mutations in [[PARK7|DJ-1]] ([http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=602533 OMIM 602533]) | |||
|- | |||
| ''PARK8'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=607060 OMIM #607060] || 12q12 || caused by mutations in [[LRRK2]] which codes for the protein [[dardarin]]. ''In vitro'', mutant LRRK2 causes protein aggregation and cell death, possibly through an interaction with parkin.<ref>{{cite journal | author=Smith WW ''et al.'' | title=Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration | journal=[[Proceedings of the National Academy of Sciences of the United States of America]] | year=2005 | volume=102 | issue=51 | pages= 18676–18681 | url=http://www.pnas.org/cgi/content/abstract/102/51/18676 | pmid = 16352719}}</ref> LRRK2 mutations, of which the most common is G2019S, cause autosomal dominant Parkinson disease, with a [[penetrance]] of nearly 100% by age 80.<ref>{{cite journal |author=Kachergus J, Mata IF, Hulihan M, ''et al'' |title=Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations |journal=Am. J. Hum. Genet. |volume=76 |issue=4 |pages=672-80 |year=2005 |pmid=15726496 |doi=10.1086/429256}}</ref> G2019S is the most common known genetic cause of Parkinson disease, found in 1-6% of U.S. and European PD patients.<ref>{{cite journal |author=Brice A |title=Genetics of Parkinson's disease: LRRK2 on the rise |journal=Brain |volume=128 |issue=Pt 12 |pages=2760-2 |year=2005 |url=http://brain.oxfordjournals.org/cgi/content/extract/128/12/2760 |pmid=16311269 |doi=10.1093/brain/awh676}} | |||
</ref> It is especially common in Ashkenazi Jewish patients, with a prevalence of 29.7% in familial cases and 13.3% in sporadic.<ref>{{cite journal | author = Ozelius L, Senthil G, Saunders-Pullman R, ''et al'' | title = LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. | journal = N Engl J Med | volume = 354 | issue = 4 | pages = 424-5 | year = 2006 | pmid = 16436782}}</ref> | |||
|- | |||
| ''PARK9'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=606693 OMIM #606693] || 1p36 || Caused by mutations in the ''ATP13A2'' gene, and also known as Kufor-Rakeb Syndrome. PARK9 may be allelic to PARK6. | |||
|- | |||
| ''PARK10'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=606852 OMIM %606852] || 1p || - | |||
|- | |||
| ''PARK11'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=607688 OMIM %607688] || 2q36-37 || However, this gene locus has conflicting data, and may not have significance. | |||
|- | |||
| ''PARK12'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=300557 OMIM %300557] || Xq21-q25 || - | |||
|- | |||
| ''PARK13'' || [http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=610297 OMIM #610297] || 2p12 || Caused by mutations in the ''HTRA2'' ([[HtrA serine peptidase 2]]) gene. | |||
|} | |||
==References== | ==References== | ||
Revision as of 21:03, 30 August 2012
|
Parkinson's disease Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Parkinson's disease pathophysiology On the Web |
|
American Roentgen Ray Society Images of Parkinson's disease pathophysiology |
|
Risk calculators and risk factors for Parkinson's disease pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
Pathophysiology
In the brain the direct pathway facilitates movement and the indirect pathway inhibits movement, thus the loss of these cells leads to a hypokinetic movement disorder. The lack of dopamine results in increased inhibition of the ventral lateral nucleus of the thalamus, which sends excitatory projections to the motor cortex, thus leading to hypokinesia.
There are four major dopamine pathways in the brain; the nigrostriatal pathway, referred to above, mediates movement and is the most conspicuously affected in early Parkinson's disease. The other pathways are the mesocortical, the mesolimbic, and the tuberoinfundibular. These pathways are associated with, respectively: volition and emotional responsiveness; desire, initiative, and reward; and sensory processes and maternal behavior. Disruption of dopamine along the non-striatal pathways likely explains much of the neuropsychiatric pathology associated with Parkinson's disease.
The mechanism by which the brain cells in Parkinson's are lost may consist of an abnormal accumulation of the protein alpha-synuclein bound to ubiquitin in the damaged cells. The alpha-synuclein-ubiquitin complex cannot be directed to the proteosome. This protein accumulation forms proteinaceous cytoplasmic inclusions called Lewy bodies. Latest research on pathogenesis of disease has shown that the death of dopaminergic neurons by alpha-synuclein is due to a defect in the machinery that transports proteins between two major cellular organelles — the endoplasmic reticulum (ER) and the Golgi apparatus. Certain proteins like Rab1 may reverse this defect caused by alpha-synuclein in animal models.[1]
Excessive accumulations of iron, which are toxic to nerve cells, are also typically observed in conjunction with the protein inclusions. Iron and other transition metals such as copper bind to neuromelanin in the affected neurons of the substantia nigra. So, neuromelanin may be acting as a protective agent. Alternately, neuromelanin (an electronically active semiconductive polymer) may play some other role in neurons.[2] That is, coincidental excessive accumulation of transition metals, etc. on neuromelanin may figure in the differential dropout of pigmented neurons in Parkinsonism. The most likely mechanism is generation of reactive oxygen species.[3]
Iron induces aggregation of synuclein by oxidative mechanisms.[4] Similarly, dopamine and the byproducts of dopamine production enhance alpha-synuclein aggregation. The precise mechanism whereby such aggregates of alpha-synuclein damage the cells is not known. The aggregates may be merely a normal reaction by the cells as part of their effort to correct a different, as-yet unknown, insult. Based on this mechanistic hypothesis, a transgenic mouse model of Parkinson's has been generated by introduction of human wild-type α-synuclein into the mouse genome under control of the platelet-derived-growth factor-β promoter.[5]
-
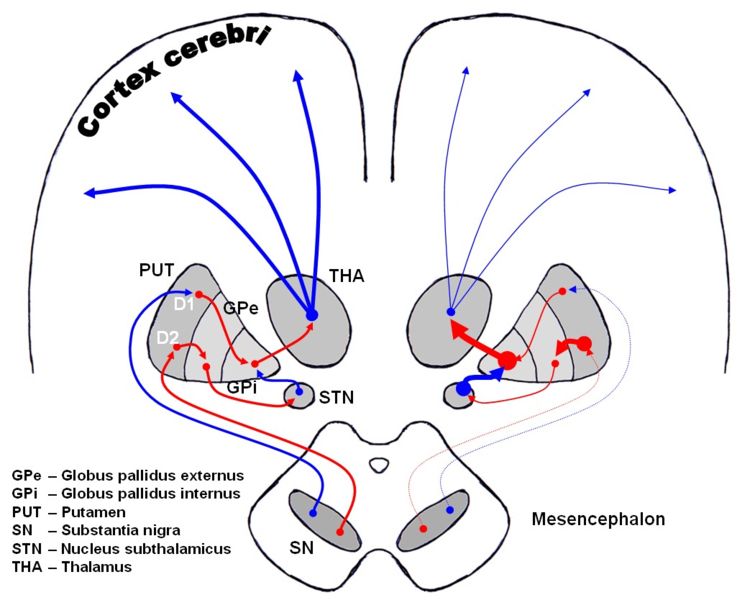
Dopaminergic pathways of the human brain in normal condition (left) and Parkinson's disease (right). Red Arrows indicate suppression of the target, blue arrows indicate stimulation of target structure.

The symptoms of Parkinson's disease result from the loss of pigmented dopamine-secreting (dopaminergic) cells, secreted by the same cells, in the pars compacta region of the substantia nigra (literally "black substance"). These neurons project to the striatum and their loss leads to alterations in the activity of the neural circuits within the basal ganglia that regulate movement, in essence an inhibition of the direct pathway and excitation of the indirect pathway.
Genetic
In recent years, a number of specific genetic mutations causing Parkinson's disease have been discovered, including in certain populations (Contursi, Italy). These account for a small minority of cases of Parkinson's disease. Somebody who has Parkinson's disease is more likely to have relatives that also have Parkinson's disease. However, this does not mean that the disorder has been passed on genetically.
Genetic forms that have been identified include:
- external links in this section are to OMIM
| Type | OMIM | Locus | Details |
| PARK1 | OMIM #168601 | 4q21 | caused by mutations in the SNCA gene, which codes for the protein alpha-synuclein. PARK1 causes autosomal dominant Parkinson disease. So-called PARK4 (OMIM #605543) is probably caused by triplication of SNCA.[6] |
| PARK2 | OMIM *602544 | 6q25.2-q27 | caused by mutations in protein parkin. Parkin mutations may be one of the most common known genetic causes of early-onset Parkinson disease. In one study, of patients with onset of Parkinson disease prior to age 40 (10% of all PD patients), 18% had parkin mutations, with 5% homozygous mutations.[7] Patients with an autosomal recessive family history of parkinsonism are much more likely to carry parkin mutations if age at onset is less than 20 (80% vs. 28% with onset over age 40).[8]Patients with parkin mutations (PARK2) do not have Lewy bodies. Such patients develop a syndrome that closely resembles the sporadic form of PD; however, they tend to develop symptoms at a much younger age. |
| PARK3 | OMIM %602404 | 2p13 | autosomal dominant, only described in a few kindreds. |
| PARK5 | OMIM +191342 | 4p14 | caused by mutations in the UCHL1 gene which codes for the protein ubiquitin carboxy-terminal hydrolase L1 |
| PARK6 | OMIM #605909 | 1p36 | caused by mutations in PINK1 (OMIM *608309) which codes for the protein PTEN-induced putative kinase 1. |
| PARK7 | OMIM #606324 | 1p36 | caused by mutations in DJ-1 (OMIM 602533) |
| PARK8 | OMIM #607060 | 12q12 | caused by mutations in LRRK2 which codes for the protein dardarin. In vitro, mutant LRRK2 causes protein aggregation and cell death, possibly through an interaction with parkin.[9] LRRK2 mutations, of which the most common is G2019S, cause autosomal dominant Parkinson disease, with a penetrance of nearly 100% by age 80.[10] G2019S is the most common known genetic cause of Parkinson disease, found in 1-6% of U.S. and European PD patients.[11] It is especially common in Ashkenazi Jewish patients, with a prevalence of 29.7% in familial cases and 13.3% in sporadic.[12] |
| PARK9 | OMIM #606693 | 1p36 | Caused by mutations in the ATP13A2 gene, and also known as Kufor-Rakeb Syndrome. PARK9 may be allelic to PARK6. |
| PARK10 | OMIM %606852 | 1p | - |
| PARK11 | OMIM %607688 | 2q36-37 | However, this gene locus has conflicting data, and may not have significance. |
| PARK12 | OMIM %300557 | Xq21-q25 | - |
| PARK13 | OMIM #610297 | 2p12 | Caused by mutations in the HTRA2 (HtrA serine peptidase 2) gene. |
References
- ↑ "Parkinson's Disease Mechanism Discovered," HHMI Research News June 22, 2006.
- ↑ McGinness J, Corry P, Proctor P (1974). "Amorphous semiconductor switching in melanins" (Reprint). Science. 183 (127): 853–5. PMID 4359339.
- ↑ Jenner P (1998). "Oxidative mechanisms in nigral cell death in Parkinson's disease". Mov Disord. 13 Suppl 1: 24–34. PMID 9613715.
- ↑ Kaur D, Andersen J (2002). "Ironing out Parkinson's disease: is therapeutic treatment with iron chelators a real possibility?" (PDF). Aging Cell. 1 (1): 17–21. PMID 12882349.
- ↑ Masliah E, Rockenstein E, Veinbergs I; et al. (2000). "Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders". Science. 287 (5456): 1265–9. PMID 10678833.
- ↑ Singleton AB, Farrer M, Johnson J; et al. (2003). "alpha-Synuclein locus triplication causes Parkinson's disease". Science. 302 (5646): 841. doi:10.1126/science.1090278. PMID 14593171.
- ↑ Poorkaj P; et al. (2004). "parkin mutation analysis in clinic patients with early-onset Parkinson's disease". American Journal of Medical Genetics Part A. 129A (1): 44&ndash, 50.
- ↑ Ebba Lohmann; et al. (2003). "How much phenotypic variation can be attributed to parkin genotype?". Annals of Neurology. 54 (2): 176&ndash, 185. PMID 12891670.
- ↑ Smith WW; et al. (2005). "Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration". Proceedings of the National Academy of Sciences of the United States of America. 102 (51): 18676&ndash, 18681. PMID 16352719.
- ↑ Kachergus J, Mata IF, Hulihan M; et al. (2005). "Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations". Am. J. Hum. Genet. 76 (4): 672–80. doi:10.1086/429256. PMID 15726496.
- ↑ Brice A (2005). "Genetics of Parkinson's disease: LRRK2 on the rise". Brain. 128 (Pt 12): 2760–2. doi:10.1093/brain/awh676. PMID 16311269.
- ↑ Ozelius L, Senthil G, Saunders-Pullman R; et al. (2006). "LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews". N Engl J Med. 354 (4): 424–5. PMID 16436782.