Multiple endocrine neoplasia type 2 pathophysiology: Difference between revisions
No edit summary |
No edit summary |
||
| Line 18: | Line 18: | ||
==Genetics== | ==Genetics== | ||
[[Image:autodominant2.jpg|thumb| | [[Image:autodominant2.jpg|thumb|center|500px|Autosomal dominent pattern of inheritance]] | ||
* Most cases of multiple endocrine neoplasia type 2 are inherited in an [[autosomal dominant]] pattern, which means affected people may have affected siblings and relatives in successive generations (such as parents and children). An affected person usually has one parent with the condition. Some cases, however, result from new mutations in the [[RET proto-oncogene]] gene. These cases occur in people with no history of the disorder in their family. | * Most cases of multiple endocrine neoplasia type 2 are inherited in an [[autosomal dominant]] pattern, which means affected people may have affected siblings and relatives in successive generations (such as parents and children). An affected person usually has one parent with the condition. Some cases, however, result from new mutations in the [[RET proto-oncogene]] gene. These cases occur in people with no history of the disorder in their family. | ||
[[File:RET kinase domain.jpg|thumb|center|500px| | [[File:RET kinase domain.jpg|thumb|center|500px|RET kinase domain]] | ||
* Germline mutations are responsible for sporadic MEN 2, while mutations in the cysteine residues in the exons of the RET protein product are common in familial MEN 2. | * Germline mutations are responsible for sporadic MEN 2, while mutations in the cysteine residues in the exons of the RET protein product are common in familial MEN 2. | ||
* Most cases of MEN2 derive from a variation in the RET proto-oncogene, and are specific for cells of neural crest origin. | * Most cases of MEN2 derive from a variation in the RET proto-oncogene, and are specific for cells of neural crest origin. | ||
Revision as of 16:53, 26 September 2015
|
Multiple endocrine neoplasia type 2 Microchapters |
|
Differentiating Multiple endocrine neoplasia type 2 from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Multiple endocrine neoplasia type 2 pathophysiology On the Web |
|
American Roentgen Ray Society Images of Multiple endocrine neoplasia type 2 pathophysiology |
|
Multiple endocrine neoplasia type 2 pathophysiology in the news |
|
Blogs on Multiple endocrine neoplasia type 2 pathophysiology |
|
Directions to Hospitals Treating Multiple endocrine neoplasia type 2 |
|
Risk calculators and risk factors for Multiple endocrine neoplasia type 2 pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Development of multiple endocrine neoplasia type 2 is the result of genetic mutations. Gene involved in the pathogenesis of multiple endocrine neoplasia type 2 include RET gene.
Pathogenesis
- The common feature among the three sub-types of MEN2 is a high propensity to develop medullary thyroid carcinoma.
Multiple endocrine neoplasia type 2A
- Multiple endocrine neoplasia type 2 (MEN2) (also known as "Pheochromocytoma and amyloid producing medullary thyroid carcinoma",[1] "PTC syndrome,"[1] and "Sipple syndrome"[1]) is a group of medical disorders associated with tumors of the endocrine system.
- They generally occur in endocrine organs (e.g. thyroid, parathyroid, and adrenals), but may also occur in endocrine tissues of organs not classically thought of as endocrine.[2]
- Although many different types of hormone-producing tumors are associated with multiple endocrine neoplasia, the most common manifestation is a form of thyroid cancer called medullary thyroid carcinoma. This tumor secretes an inactive hormone called calcitonin. Many people with this disorder also develop pheochromocytoma, which is a tumor of the adrenal glands (located above each kidney) that can cause dangerously high blood pressure. In addition, overactivity of the parathyroid gland (hyperparathyroidism) occurs in some cases of multiple endocrine neoplasia type 2. Hyperparathyroidism disrupts the normal balance of calcium in the blood, which can lead to kidney stones, thinning of bones, weakness, and fatigue.
- In MEN2A primary hyperparathyroidism occurs in only 10–30% and is usually diagnosed after the third decade of life. It can occur in children but this is rare. It may be the sole clinical manifestation of this syndrome but this is unusual.
- MEN2A associates medullary thyroid carcinoma with pheochromocytoma in about 20–50% of cases and with primary hyperparathyroidism in 5–20% of cases.
- In familial isolated medullary thyroid carcinoma the other components of the disease are absent.
Multiple Endocrine Neoplasia type 2B
- Multiple endocrine neoplasia type 2B (also known as MEN2B, Mucosal neuromata with endocrine tumors, Multiple endocrine neoplasia type 3, and Wagenmann–Froboese syndrome[1]) is a genetic disease that causes multiple tumors on the mouth, eyes, and endocrine glands. It is the most severe type of multiple endocrine neoplasia,[3] differentiated by the presence of benign oral and submucosal tumors in addition to endocrine malignancies.
- MEN2B associates medullary thyroid carcinoma with pheochromocytoma in 50% of cases, with marfanoid habitus and with mucosal and digestive neurofibromatosis.
Genetics

- Most cases of multiple endocrine neoplasia type 2 are inherited in an autosomal dominant pattern, which means affected people may have affected siblings and relatives in successive generations (such as parents and children). An affected person usually has one parent with the condition. Some cases, however, result from new mutations in the RET proto-oncogene gene. These cases occur in people with no history of the disorder in their family.

- Germline mutations are responsible for sporadic MEN 2, while mutations in the cysteine residues in the exons of the RET protein product are common in familial MEN 2.
- Most cases of MEN2 derive from a variation in the RET proto-oncogene, and are specific for cells of neural crest origin.
- The protein produced by the RET gene plays an important role in the TGF-beta (transforming growth factor beta) signaling system. Because the TGF-beta system operates in nervous tissues throughout the body, variations in the RET gene can have effects in nervous tissues throughout the body.
- The RET protooncogene is a 21-exon gene and encodes for a tyrosine kinase transmembrane receptor located on chromosome 10q11.2.[4]
- Four different ligands have so far been recognized: the glial cell-line derived neutrophilic factor (GDNF), neurturin (NTN), persepin (PNS) and artemin (ART). The interaction is mediated by a ligand-specific coreceptor (e.g., the GFRα-1 is the co-receptor for the GDNF).
- The receptor is composed of an extracellular domain (EC), with a distal cadherin-like region and a juxtamembrane cystein-rich region, a transmembrane domain (TM) and an intracellular domain with tyroisine-kinase activity (TK).
Table below summerizes specific RET codons and their functions.[5]
| Mutation location | RET codons | Function of wild type codon | Mutated effects | Phenotype |
|---|---|---|---|---|
| Extracellular cystine rich location |
|
Helps to form teritiary structure with the help of disulfide bonds | Alteration in protein folding and maturation | MEN 2A and FMTC |
| c634 | Formation of intramolecular disulfide bonds | Ligand independant dimerization of receptor molecules | MEN 2A | |
| Intracellular tyrosine kinase domain |
|
Terminal lobe of RET kinase | Affects ATP binding and interlobe flexibility | MEN 2A and FMTC |
|
Close proximity with ATP binding site | Alters interactions within the region | FMTC | |
|
Gatekeeper residue that regulates access to ATP binding site | Alters interactions within the region | FMTC | |
|
C terminal lobe of kinase | Alters activation of loop conformation | MEN 2A and FMTC | |
|
Situated next to activated loop | Local conformational change | MEN 2B | |
|
Substrate binding pocket of the kinase | Alters protein conformation | MEN 2B |
- MEN2 generally results from a gain-of-function variant of a RET gene. Other diseases, such as Hirschsprung disease, result from loss-of-function variants.
- When inherited, multiple endocrine neoplasia type 2 is transmitted in an autosomal dominant pattern, which means affected people have one affected parent, and possibly-affected siblings and children. Some cases, however, result from spontaneous new mutations in the RET gene. These cases occur in people with no family history of the disorder. In MEN2B, for example, about half of all cases arise as spontaneous new mutations.
- Activating germline point mutations of the RET proto-oncogene are causative events in MEN 2A, MEN 2B, and FMTC. RET mutations have been found to be widely distributed not only among the 5 cysteine codons 609, 611, 618, 620, and 634 but also in other noncysteine codons, such as codon 804 in exon 14, codon 883 in exon 15, and others.
- The following figue depicts the structure and mutation of RET receptor.[5]

RET Activation
- Dimerization of RET gene mediated through formation of multicomponent complex. RET is activated by binding both a soluable ligand and a a non signaling extracellular co-receptor. RET activation leads to phosphorylation of multiple intracellular tyrosine kinase.
Associated Conditions
Some of the diseases specific to the genes of multiple endocrine neoplasia type 2 are as follows.[5]
| Subtype | Associated diseases |
|---|---|
| MEN2A | Cutaneous lichen amyloidosis, Hirschsprung disease |
| MEN2B | Ganglioneuromatosis, marfanoid habitus |
| FMTC | Rare diseases |
Gross Pathology
-
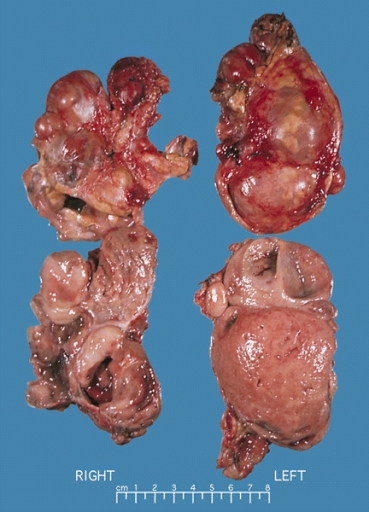
ADRENAL GLAND: BILATERAL PHEOCHROMOCYTOMA Cross section of bilateral pheochromocytomas from a 30-year-old man with MEN syndrome type IIa. The right adrenal tumor weighed 168 g and the left 220 g. Note the distinct multinodular, multicentric pattern of growth on both sides
-
![Pheochromocytoma, From the case [6]](/images/3/38/Pheochromocytoma_03.jpeg)
Pheochromocytoma, From the case [6]
-
![Pheochromocytoma, From the case [7]](/images/6/6a/Pheochromocytoma_04.JPG)
Pheochromocytoma, From the case [7]

![Pheochromocytoma, From the case [6]](/index.php/File:Pheochromocytoma_03.jpeg)
![Pheochromocytoma, From the case [7]](/index.php/File:Pheochromocytoma_04.JPG)
Microscopic Pathology
Medullary Carcinoma of Thyroid
- Nests of C cells invading the basement membrane and infiltrating thyroid follicles.
-
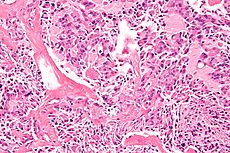
High magnification micrograph of medullary thyroid carcinoma, abbreviated MTC. H&E stain. MTC can be remembered by the 3 Ms:aMyloid. Median node dissection. MEN 2A & MEN 2B. It typically stains with: Calcitonin. CEA. Chromogranin A. Multiple endocrine neoplasia 2A: Medullary thyroid carcinoma
-
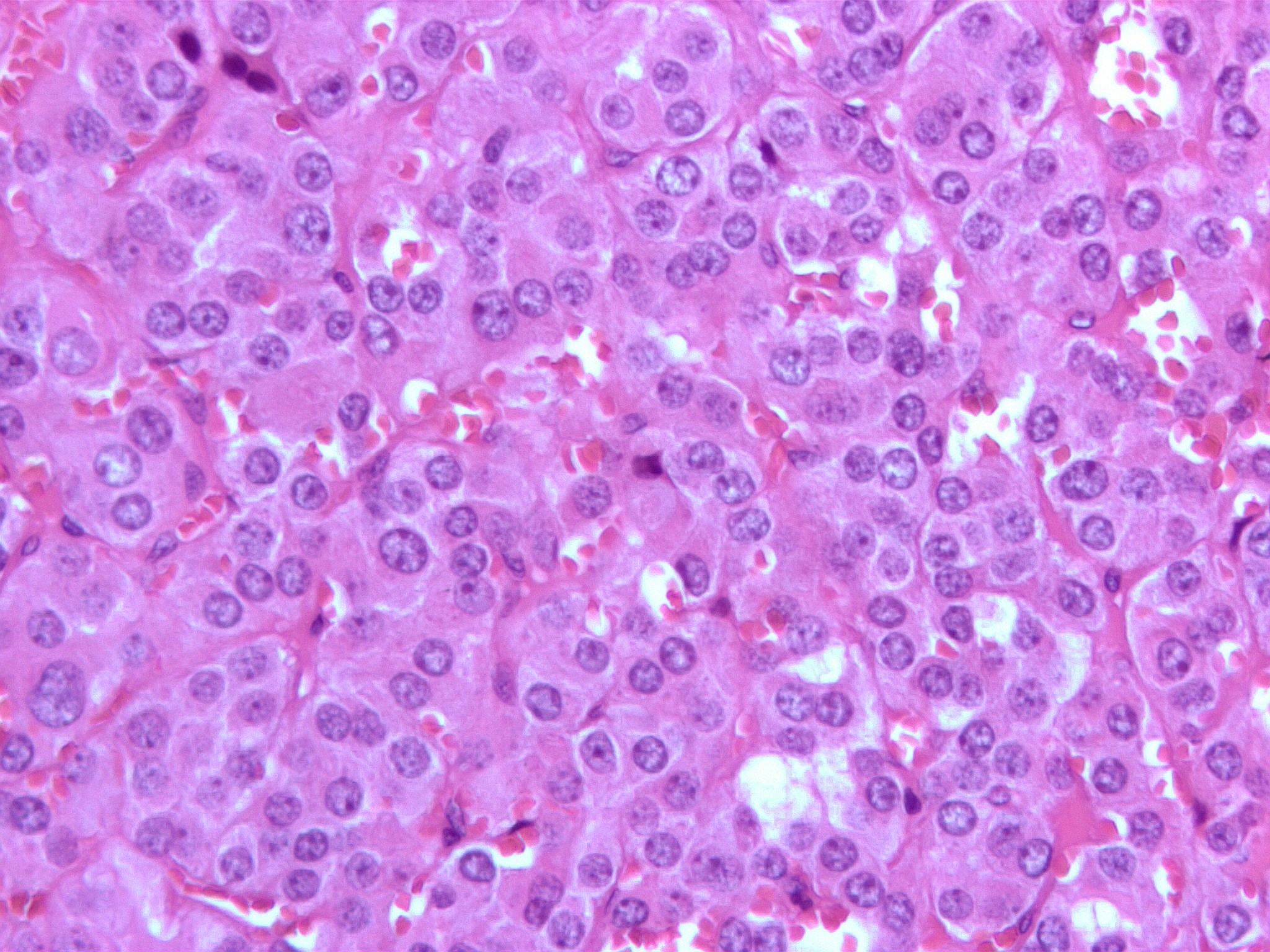
Micrograph of a pheochromocytoma (at high magnification) showing the characteristic stippled (finely granular) chromatin. The chromatin pattern is sometimes referred to as "salt-and-pepper" chromatin
-
![Pheochromocytoma, Case courtesy of Dr Andrew Ryan, [8]](/images/d/d9/Pheochromocytoma_histology.jpg)
Pheochromocytoma, Case courtesy of Dr Andrew Ryan, [8]


![Pheochromocytoma, Case courtesy of Dr Andrew Ryan, [8]](/index.php/File:Pheochromocytoma_histology.jpg)
References
- ↑ 1.0 1.1 1.2 1.3 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- ↑ Moline J, Eng C. (2011). "Multiple endocrine neoplasia type 2: an overview". Genet Med. 9 (13): 755–64. doi:10.1097/GIM.0b013e318216cc6d. PMID 21552134.
- ↑ Carlson KM, Bracamontes J, Jackson CE, et al. (December 1994). "Parent-of-origin effects in multiple endocrine neoplasia type 2B". Am. J. Hum. Genet. 55 (6): 1076–82. PMC 1918453. PMID 7977365.
- ↑ Schmoldt A, Benthe HF, Haberland G (1975). "Digitoxin metabolism by rat liver microsomes". Biochem Pharmacol. 24 (17): 1639–41. PMID http://dx.doi.org/10.1155/2012/705036 Check
|pmid=value (help). - ↑ 5.0 5.1 5.2 Schmoldt A, Benthe HF, Haberland G (1975). "Digitoxin metabolism by rat liver microsomes". Biochem Pharmacol. 24 (17): 1639–41. PMID http://dx.doi.org/10.6061/clinics/2012(Sup01)14 Check
|pmid=value (help). - ↑ "http://radiopaedia.org/cases/10816">rID: 10816
- ↑ "http://radiopaedia.org/cases/10816">rID: 10816
- ↑ "http://radiopaedia.org/">Radiopaedia.org</a>. From the case <a href="http://radiopaedia.org/cases/22683">rID: 22683