Androgen insensitivity syndrome pathophysiology
|
Androgen insensitivity syndrome Microchapters |
|
Differentiating Androgen insensitivity syndrome from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Androgen insensitivity syndrome pathophysiology On the Web |
|
American Roentgen Ray Society Images of Androgen insensitivity syndrome pathophysiology |
|
Directions to Hospitals Treating Androgen insensitivity syndrome |
|
Risk calculators and risk factors for Androgen insensitivity syndrome pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief:
Overview
Androgen insensitivity syndrome results from mutations of the gene encoding the androgen receptor. AIS involves variable degree of undervirilization and/or infertility in XY persons of either sex.
Pathophysiology
- Androgen insensitivity syndrome results from mutations of the gene encoding the androgen receptor. It has also been called androgen resistance in the medical literature.
- The nature of the resulting problem varies according to the structure and sensitivity of the abnormal receptor.
- Most of the forms of AIS involve variable degrees of undervirilization and/or infertility in XY persons of either sex. A woman with complete androgen insensitivity syndrome (CAIS) has a nearly normal female body despite a 46XY karyotype and undescended testes, a condition termed testicular feminization in the past.
Early developmental/embryological aspect
- In 8th to 10th week of gestation, male gonads start to produce testosterone, dihydrotestosterone(DHT), and Mullerian inhibiting factor(MIF), the hormones important for the male development.
- Normal production of the hormone levels occurs, but improper functioning of androgen receptors for testosterone and dihydrotestosterone(DHT) is observed.
- Concurrently, when exposed to the Mullerian inhibiting factor, it causes internal male development, while unopposed testicular estrogen results in the development of external female genitalia.
Prenatal effects of testosterone in 46,XY fetus
- In a normal fetus with a 46,XY karyotype, the presence of the SRY gene induces testes to form on the genital ridges in the fetal abdomen a few weeks after conception. By 6 weeks of gestation, genital anatomies of XY and XX fetuses are still indistinguishable, consisting of a tiny underdeveloped button of tissue able to become a phallus, and a urogenital midline opening flanked by folds of skin able to become either labia or scrotum. By the 7th week, fetal testes begin to produce testosterone and release it into the blood.
- Directly and as DHT, testosterone acts on the skin and tissues of the genital area and by 12 weeks of gestation, has produced a recognizable male, with a growing penis with a urethral opening at the tip, and a perineum fused and thinned into a scrotum, ready for the testes. Evidence suggests that this "remodelling" of the genitalia can only occur during this period of fetal life; if not complete by about 13 weeks, no amount of testosterone later will move the urethral opening or close a vagina-like opening.
- For the remainder of gestation, the principal known effect of testosterone and DHT is continued growth of the penis and internal wolffian derivatives (part of prostate, epididymis, seminal vesicles, and vas deferens).
Early postnatal effects of testosterone in 46,XY infants
- Testosterone levels are low at birth but rise within weeks, remaining at normal male pubertal levels for about 2 months before declining to the low, barely detectable childhood levels. The biological function of this rise is unknown. Animal research suggests a contribution to brain differentiation.
Pubertal effects of testosterone in 46,XY children
- At puberty, many of the early physical changes in both sexes are androgenic (adult-type body odor, increased oiliness of skin and hair, acne, pubic hair, axillary hair, fine upper lip and sideburn hair).
- As puberty progresses, later secondary sex characteristics in males are nearly entirely due to androgens (continued growth of the penis, maturation of spermatogenic tissue and fertility, beard, deeper voice, masculine jaw and musculature, body hair, heavier bones). In males, the major pubertal changes attributable to estradiol are growth acceleration, epiphyseal closure, termination of growth, and (if it occurs) gynecomastia.
Genetics
| Molecular Genetic Testing Used in Androgen Insensitivity Syndrome | ||
| Gene | Test Method | Proportion of 46,XY Probands w/a Pathogenic Variant 2 Detectable by This Method |
|---|---|---|
| AR | Sequence analysis |
|
| Gene-targeted deletion/duplication analysis |
| |
- Family history of other affected individuals related to each other in a pattern consistent with X-linked inheritance. “Other affected family members” refers to: [1]
- Affected 46,XY individuals
- Manifesting heterozygous females (46,XX).
- About 10% of heterozygous females have asymmetric distribution and sparse or delayed growth of pubic and/or axillary hair.
- Absence of a family history of AIS or suggestive features of AIS does not preclude the diagnosis.

- Because the Androgen Insensitivity Syndrome gives rise to ambiguity between the genetic and the phenotypic gender, we will use the convention 46,XY to designate a genotypic male, and 46,XX to designate a genotypic female. By this convention, a person with Androgen Insensitivity Syndrome is a 46,XY but a phenotypic female.
- The Androgen Insensitivity Syndrome has been linked to mutations in AR, the gene for the human Androgen Receptor, located at Xq11-12 (i.e. on the X chromosome). Thus, it is an X-linked recessive trait, causing minimal or no effects in 46,XX people.

- However, 46,XX women with a single mutated copy of the AR gene can be "carriers" of AIS, and their 46,XY children (male) will have a 50% chance of having the syndrome. As in some other X-linked recessive conditions, carrier mothers may display some minor traits of the condition: AIS carriers often have reduced axillary and pubic hair, and reduced normal adolescent acne.
Associated Conditions
Gross Pathology
- Complete androgen insensitivity syndrome in a 30 years old woman who presented primary amenorrhea.[2]. [3]
-
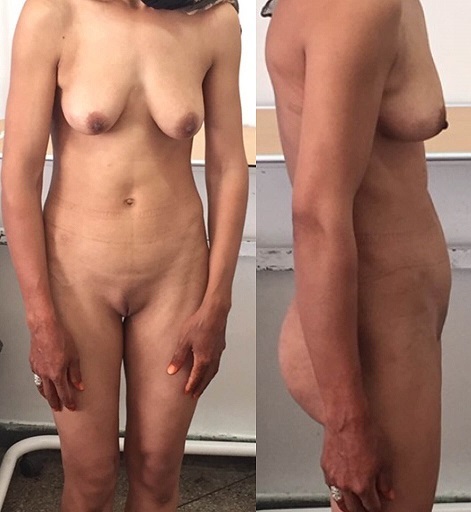
Front and side view of the patient
-
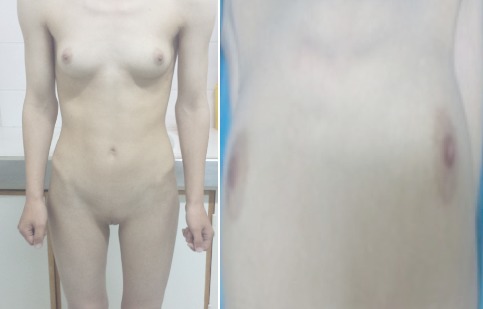
Normal female morphotype but absence of pubic and axillary hair
-
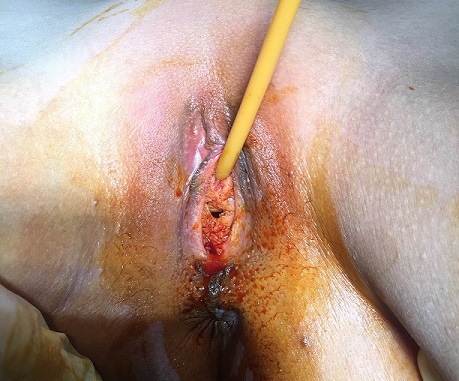
Clinical aspect of the vagina
-
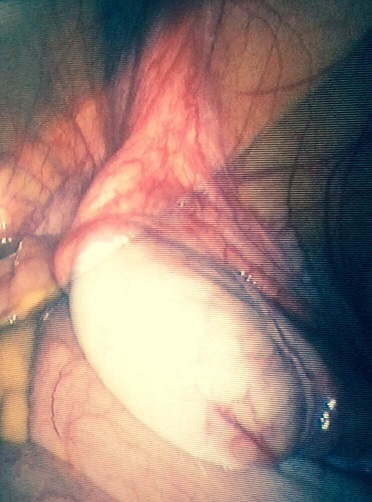
Intra- abdominal testes - Laparoscopic aspect
-
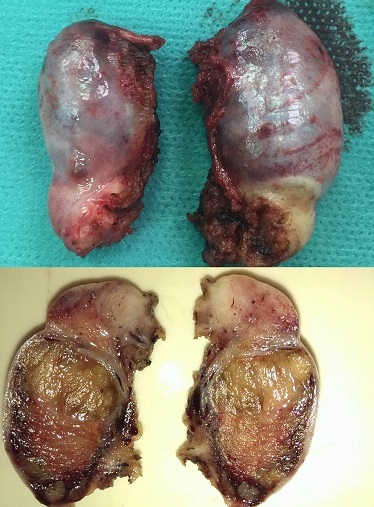
The excised testis - Macroscopic aspect





Microscopic Pathology
- Histopathology shows two testes with atrophic seminiferous tubules containing only Sertoli cells, associated to a Leydig cells hyperplasia.[3]
- On histological examination, the well-limited nodule circumscribed by a thin capsule consists of atrophic Servolian tubes with a very small interstitial tissue with rare Leydig cells. This nodule corresponds to a well differentiated tumor with Sertoli-Leydig cells. [2]
-
![Testes - Atrophy of the seminiferous tubules - Histopathological aspect, HE staining.[2]](/images/b/b5/AIS_-_Testes_-_atrophy_of_the_seminiferous_tubules_-_Histopathological_aspect%2C_HE_staining.jpg)
Testes - Atrophy of the seminiferous tubules - Histopathological aspect, HE staining.[2]
-
![testicular parenchyma of lobulated architecture, made of seminiferous tubes of atrophic appearance; B) these tubes are lined with Sertolia cells, with no obvious signs of spermatogenesis the interstitium is fibrous with rare clusters of Leydig cells.[3]](/images/8/8a/AIS_-_testicular_parenchyma.jpg)
testicular parenchyma of lobulated architecture, made of seminiferous tubes of atrophic appearance; B) these tubes are lined with Sertolia cells, with no obvious signs of spermatogenesis the interstitium is fibrous with rare clusters of Leydig cells.[3]
![Testes - Atrophy of the seminiferous tubules - Histopathological aspect, HE staining.[2]](/index.php/File:AIS_-_Testes_-_atrophy_of_the_seminiferous_tubules_-_Histopathological_aspect,_HE_staining.jpg)
![testicular parenchyma of lobulated architecture, made of seminiferous tubes of atrophic appearance; B) these tubes are lined with Sertolia cells, with no obvious signs of spermatogenesis the interstitium is fibrous with rare clusters of Leydig cells.[3]](/index.php/File:AIS_-_testicular_parenchyma.jpg)
References
- ↑ Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean L, Bird TD, Ledbetter N, Mefford HC, Smith R, Stephens K, Gottlieb B, Trifiro MA. PMID 20301602. Vancouver style error: initials (help); Missing or empty
|title=(help) - ↑ 2.0 2.1 2.2 Souhail R, Amine S, Nadia A, Tarik K, Khalid EK, Abdellatif K, Ahmed A (2016). "Complete androgen insensitivity syndrome or testicular feminization: review of literature based on a case report". Pan Afr Med J. 25: 199. doi:10.11604/pamj.2016.25.199.10758. PMC 5326263. PMID 28270903.
- ↑ 3.0 3.1 3.2 Lachiri B, Hakimi I, Boudhas A, Guelzim K, Kouach J, Oukabli M, Rahali DM, Dehayni M (2015). "[Complete androgen insensitivity syndrome: report of two cases and review of literature]". Pan Afr Med J (in French). 20: 400. doi:10.11604/pamj.2015.20.400.6760. PMC 4524922. PMID 26301004.