Acute myeloid leukemia pathophysiology
| https://https://www.youtube.com/watch?v=itkRVTqfPsE%7C350}} |
|
Acute myeloid leukemia Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Acute myeloid leukemia pathophysiology On the Web |
|
American Roentgen Ray Society Images of Acute myeloid leukemia pathophysiology |
|
Risk calculators and risk factors for Acute myeloid leukemia pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Raviteja Guddeti, M.B.B.S. [2] Carlos A Lopez, M.D. [3]
Overview
Acute myeloid leukemia arises from myeloblasts, which are hematologic white cells that are normally involved in hematopoiesis. Genetic translocations involved in the pathogenesis of acute myeloid leukemia include translocations between chromosome 8 and 21 t(8;21) and translocations between chromosome 15 and 17 t(15;17). Inversions in the chromosomal translocations in chromosome 16 inv(16) are also involved in the pathogenesis of acute myeloid leukemia.
Pathophysiology
- The malignant cell in acute myeloid leukemia is the myeloblast. In normal hematopoiesis, the myeloblast is an immature precursor of myeloid white blood cells; a normal myeloblast will gradually mature into a mature white blood cell. However, in acute myeloid leukemia a single myeloblast accumulates genetic changes which "freeze" the cell in its immature state and prevent differentiation.[1] Such a mutation alone does not cause leukemia; however, when such a "differentiation arrest" is combined with other mutations which disrupt genes controlling proliferation, the result is the uncontrolled growth of an immature clone of cells, leading to the clinical entity of acute myeloid leukemia.[2]
- Much of the diversity and heterogeneity of acute myeloid leukemia stems from the fact that leukemic transformation can occur at a number of different steps along the differentiation pathway.[3]
- Modern classification schemes for acute myeloid leukemia recognize that the characteristics and behavior of the leukemic cell (and the leukemia) may depend on the stage at which differentiation was halted.
Normal Hematopoeisis
- Hematopoeisis is tightly regulated under physiological conditions via a number of lineage-specific growth factor and lineage-specific growth signalling pathways.
- The differentiation of myeloid stem cells into mature myelocytes is controlled by lineage-specific transcription factors that regulate the expression of lineage-specific genes.
- Normal hematopoeisis is dependent upon specific growth factor receptors. The two key growth factor receptors involved are as following:
- Growth factor receptors with intrinsic tyrosine kinase activity (expressed on CD34+ hematopoietic progenitor cells, examples include, receptors for the platelet-derived growth factors (PDGFs) PDGFR A and B, the receptor for M-CSF, Fms, and the receptors for KL (SCF) and FL, Kit and Flt3)
- Growth factor receptors without intrinsic tyrosine kinase activity and depend upon intracellular kinases of the Src and JAK families (examples include, the JAK-STAT pathway)
Role Altered Signal Transduction and Autonomous Proliferaion (Protein Tyrosine Kinase Activation)
- Activation of tyrosine kinase receptors is followed by signal transduction via intracellular signal cascades leading to alteration of transcription within the cell nucleus.
- An important pathway that leads to cellular proliferation is the Ras-MAP Kinase pathway, where Ras is activated by binding of GTP. Ras-bound GTP in turn triggers acascade of events that finally lead to activation of serine/threonine kinases. Consequently, there is an activation of MAP kinases, which phosphorylate important transcriptional regulators of cell cycle.
- As a consequence of these, there is autonomous increased proliferation of cells.
Role of Altered Gene Expression and Differentiation Blockade
- Altered gene expression leads to autonomous cell proliferation with defect in regulatory pathways involved in cellular proliferation.
- Chromosomal translocations and point mutations. These processes play pivotal role in generating a differentiation blockade on myeloid cells.
- This results in a disruption in transcription factors..
- Transcription factors affected by chromosomal rearrangement (translocations) include:
- Core binding factor complex [t(8;21)-Runx1-MTG8 fusion; Inversion of chromosome 16-yielding the CBFβ-MYH11 fusion; t(3,21)-generating the RUNX1-EVI1 fusion]
- Retinoic acid receptor (RAR) [t(15;17), which generates the PML-RARα fusion]
- MLL protein [Activator protein of Hox gene promoters, Hox gene promoters in turn promote self-renewal of immature myeloid cells]
- Hox proteins
- Point mutations in myeloid transcription factors include:
- C/EBPα
- PU.1
Evasion of Apoptosis (Protein Tyrosine Kinase Activation)
- The increased expression of Bcl-2 pro-survival molecule plays a key role in evasion of programmed cell death in AML.
- PI 3-kinase activate the AKT serine/threonine kinase, and this kinase in turn phosphorylates BAD and releases the BCL-2
- The RUNX1-MTG8 fusion protein of AML represses the expression of p14ARF and promotes destabilization of p53 (a tumor supressor gene)
Self-Renewal
- The myeloid cells in acute myeolid leukemia have an ability to self-renew without being committed to a specific cell lineage.
- The self-renewing capacity of myeloid cells in AMLs is thought to be mediated via the following:
- Fusion of ALK tyrosine kinase with nucleophosmin protein (NPM)
- Mutated FLT3-ITD
- RUNX1-MTG8, PML-RARα , and PLZF-RARα fusions can all induce the expression of β-catenin and γ-catenin (plako-globin) proteins[4]
Genetics
Specific cytogenetic abnormalities can be found in many patients with acute myeloid leukemia; the types of chromosomal abnormalities often have prognostic significance.[5] The chromosomal translocations encode abnormal fusion proteins, usually transcription factors whose altered properties may cause the "differentiation arrest."[6] For example, in acute promyelocytic leukemia, the t(15;17) translocation produces a PML-RARα fusion protein which binds to the retinoic acid receptor element in the promoters of several myeloid-specific genes and inhibits myeloid differentiation.[7]. Acute myeloid leukemia M2 subtype is characterized by a translocation of a part of chromosome 8 to chromosome 21, written as t(8;21). On both sides of the chromosome, now containing pieces from two chromosomes, the DNA codes for different proteins. These two proteins are now being created as one single large protein, with a different effect in the body as the two proteins originally coded by the two different chromosomes. The two different proteins that are fused together are: RUNX1 and ETO.
The clinical signs and symptoms of acute myeloid leukemia result from the fact that, as the leukemic clone of cells grows, it tends to displace or interfere with the development of normal blood cells in the bone marrow.[8] This leads to neutropenia, anemia, and thrombocytopenia. The symptoms of acute myeloid leukemia are in turn often due to the low numbers of these normal blood elements. In rare cases, patients can develop a chloroma, or solid tumor of leukemic cells outside the bone marrow, which can cause various symptoms depending on its location.
Microscopic Pathology
Description of pictures according the classification of Acute myeloid leukemia system.
Acute myeloid leukemia M0 classification
- Acute myeloid leukemia M0 with lack of obvious myeloid differentiation by routine histologic examination and presence of myeloperoxidase in <3% of blasts. Morphologically, blasts are small to large with no granules or Auer rods.
- Acute myeloid leukemia-M1 with presence of more than 90% myeloblasts in blood.
- Acute myeloid leukemia-M1 peroxidase.
Acute myeloid leukemia M2 classification
- Acute myeloid leukemia-M2: Presence of granules can be noted.
- Acute myeloid leukemia-M2: Large myeloblasts with prominent nucleoli.
Acute myeloid leukemia M3 classification
- Acute myeloid leukemia-M3: Also called promyelocytic leukemia. Hypergranular morphology with most cells containing abundant large granules.
- Acute myeloid leukemia-M3: Ruptured cells are releasing their granules free onto the slide. Presence of Auer rods can be noticed.
Acute myeloid leukemia M5a and M5b classification
- Acute myeloid leukemia-M5a: >80% monoblasts in the marrow.
- Acute myeloid leukemia-M5a: large monoblasts with fine nuclear chromatin and prominent nucleoli. Note the absence of Auer rods.
- Acute myeloid leukemia-M5b: <80% monoblasts in the marrow.
Acute myeloid leukemia M7 classification
- Acute myeloid leukemia-M7: Irregular cytoplasmic border is often noted in some of the megakaryoblasts and occasionally projections resembling budding atypical platelets are present.
- Megakaryoblasts are usually medium-sized to large cells with a high nuclear-cytoplasmic ratio
- Nuclear chromatin is dense and homogeneous
- Variable basophilic cytoplasm which may be vacuolated
- An irregular cytoplasmic border is often noted in some of the megakaryoblasts and occasionally projections resembling budding atypical platelets are present
- Megakaryoblasts lack myeloperoxidase (MPO) activity and stain negatively with Sudan black B
- Clumps or granules in the cytoplasm
- PAS staining varies from negative to focal or granular positivity, to strongly positive staining
- More precise identification is by immunophenotyping or with electron microscopy (EM)
- Immunophenotyping using MoAb to megakaryocytic restricted antigen (CD41 and CD61) may be diagnostic
(Images shown below are courtesy of Melih Aktan MD., Istanbul Medical Faculty - Turkey, and Kyoto University - Japan)
-
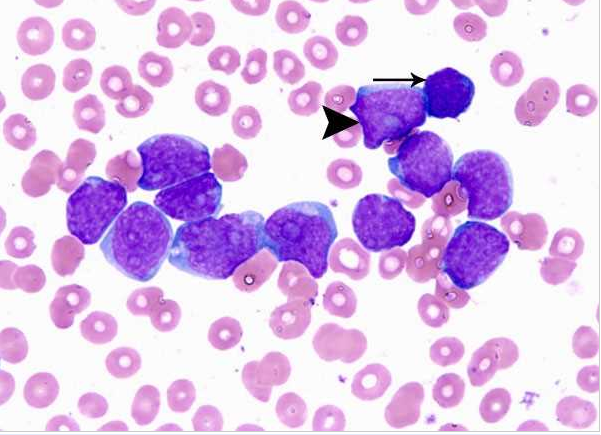
Acute myeloid leukemia-M0 - lack of obvious myeloid differentiation by routine histologic examination and presence of myeloperoxidase in <3% of blasts. Morphologically, blasts are small to large with no granules or Auer rods.
-
Acute myeloid leukemia-M1 - presence of more than 90% myeloblasts in blood.
-
Acute myeloid leukemia-M1 peroxidase.
-
Acute myeloid leukemia-M1 - more cytoplasm than M0, but still no granules.
-
Acute myeloid leukemia-M1.
-
Acute myeloid leukemia-M1.
-
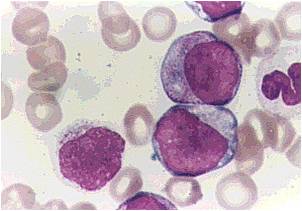
AML-M2 - presence of granules can be noted.
-
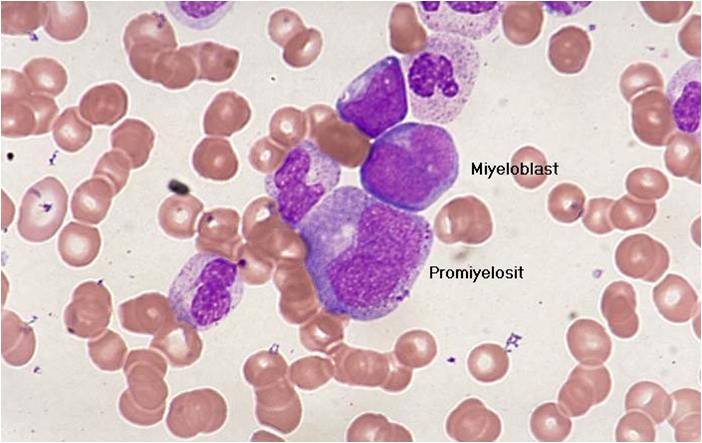
AML-M2 - large myeloblasts with prominent nucleoli.
-
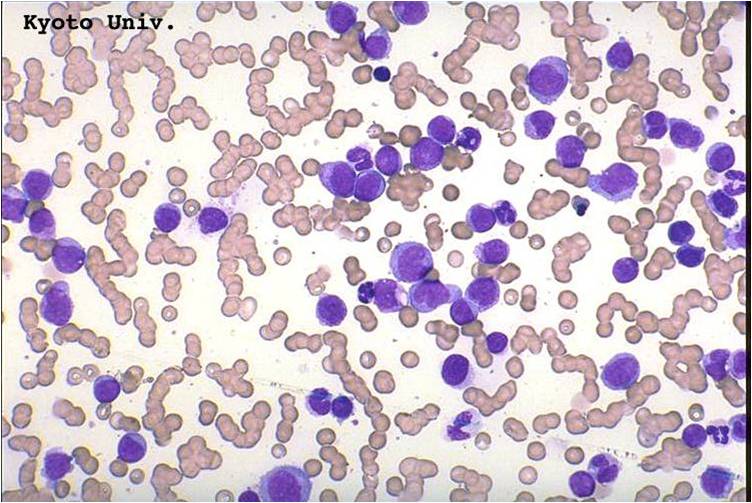
AML-M2 - maturing myeloid elements including band and segmented neutrophils in the background.
-
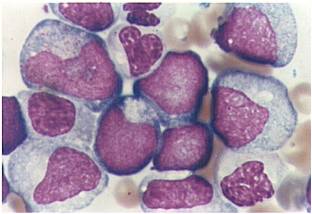
AML-M2 - presence of a few maturing myeloid elements.
-
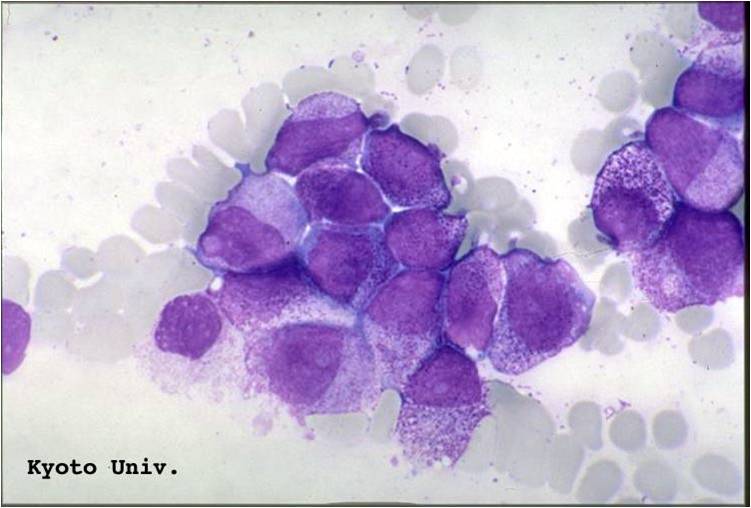
AML-M3 - also called promyelocytic leukemia. Hypergranular morphology with most cells containing abundant large granules.
-
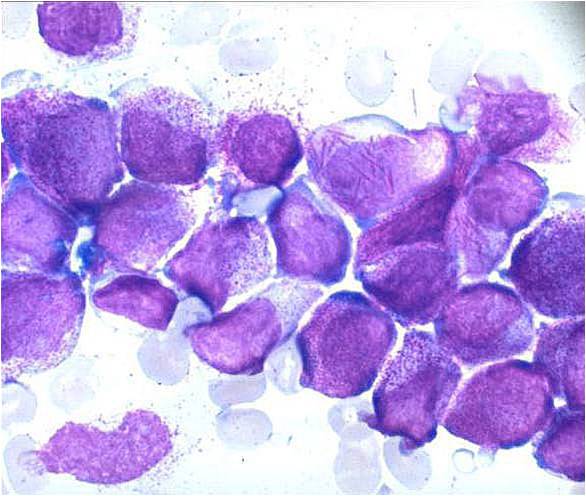
AML-M3 - ruptured cells are releasing their granules free onto the slide. Presence of Auer rods can be noticed.
-
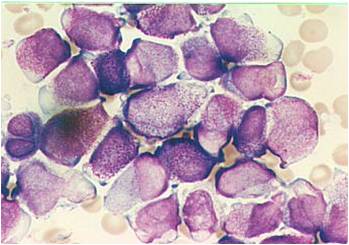
AML-M3 - presence of abundant fine granules.
-
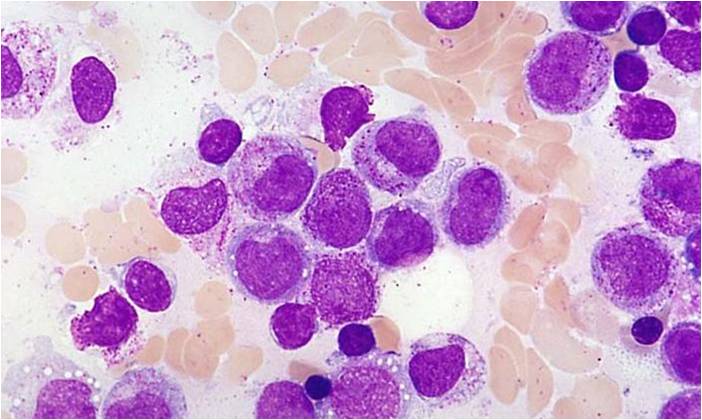
AML-M3 - polarity of cytoplasmic granulation. In many cells the granules tend to polarize toward one portion of the cytoplasm and the nucleus on the opposite side.
-
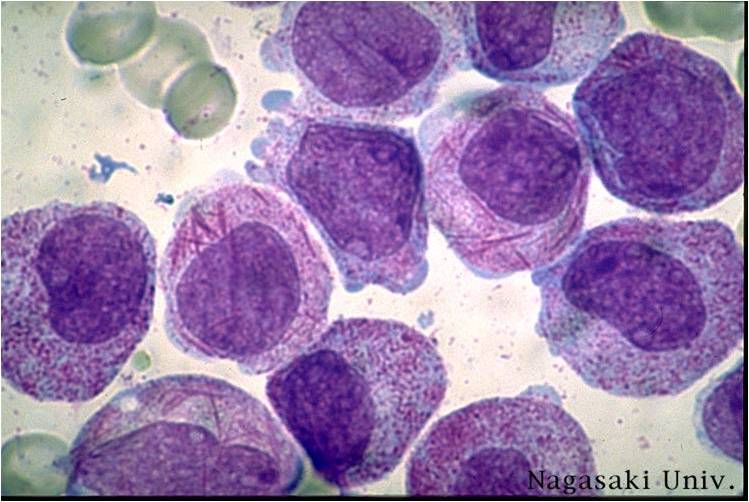
AML-M3 Auer bodies.
-
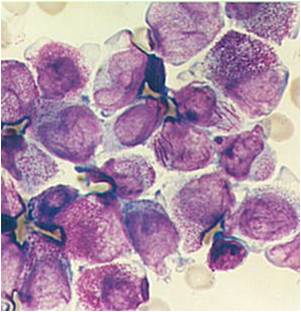
AML-M3 Auer bodies.
-
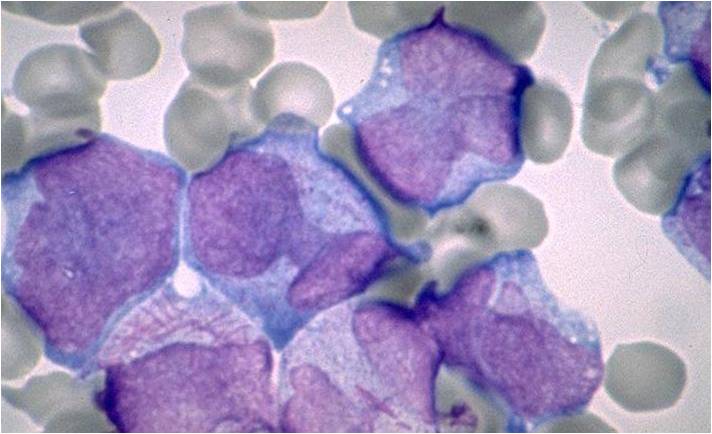
AML-M3 variation.
-
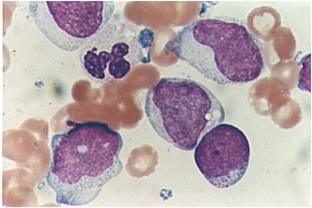
AML-M4 - large monoblasts with abundant cytoplasm and moderately to intensely basophilic. The monoblasts will have lacy nuclear chromatin and one or more predominant nucleoli.
-
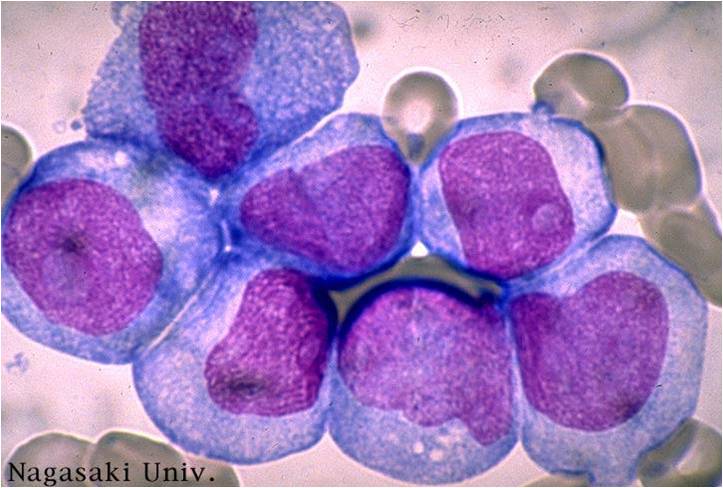
AML-M5a - >80% monoblasts in the marrow.
-
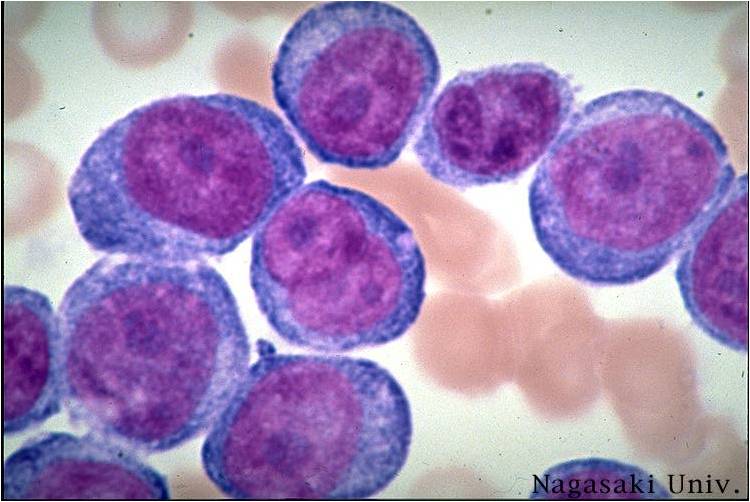
AML-M5a - large monoblasts with fine nuclear chromatin and prominent nucleoli. Note the absence of Auer rods.
-
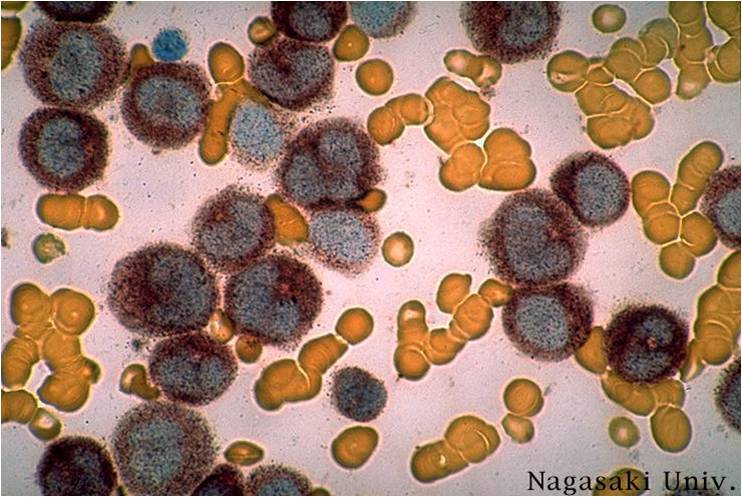
AML-M5a (alpha naphtyle acetat).
-
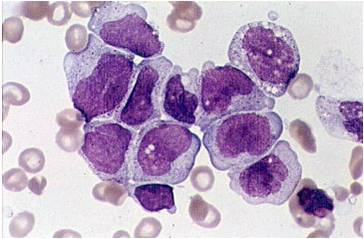
AML-M5b - <80% monoblasts in the marrow.
-
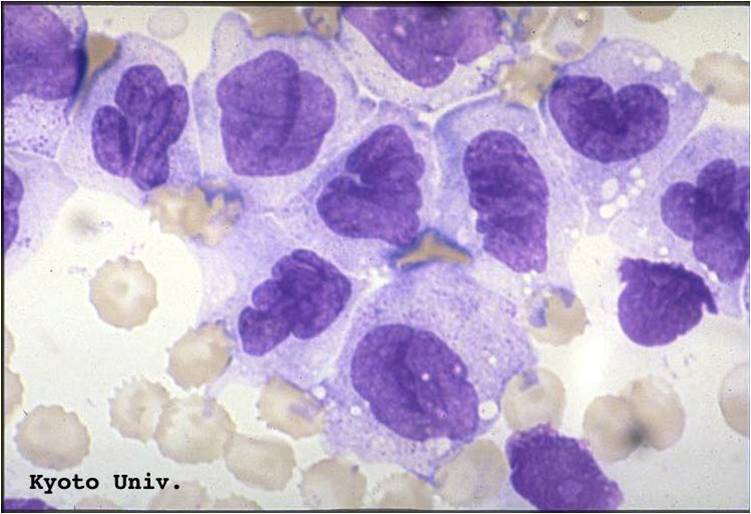
AML-M5b - moderate amounts of cytoplasm and large nuclei with fine chromatin but without prominent nucleoli. Note presence of nuclear folds and creases, which are a characteristic feature of promonocytes.
-
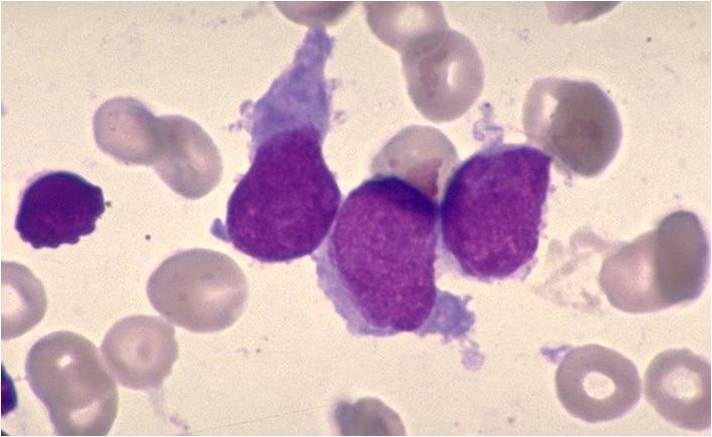
AML-M7 - irregular cytoplasmic border is often noted in some of the megakaryoblasts and occasionally projections resembling budding atypical platelets are present.
-
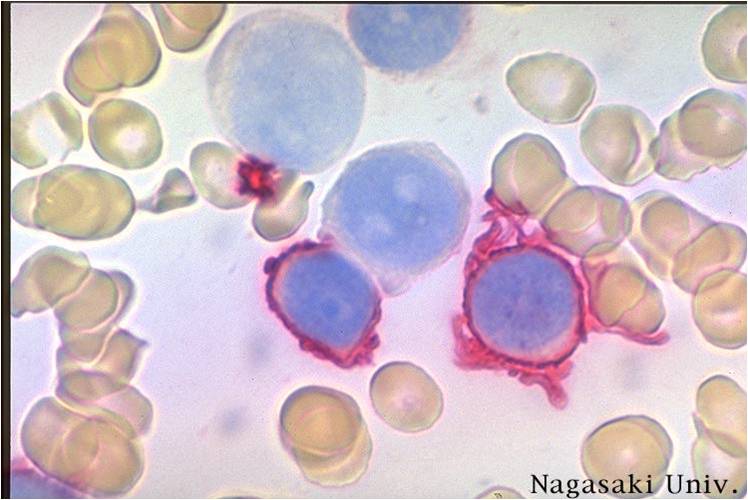
AML-M7 - megakaryoblasts are usually medium sized to large cells with a high nuclear-cytoplasmic ratio. Nuclear chromatin is dense and homogeneous.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- ↑ Fialkow PJ: Clonal origin of human tumors. Biochim Biophys Acta 1976;458:283–321. PMID 1067873
- ↑ Fialkow PJ, Janssen JW, Bartram CR: Clonal remissions in acute nonlymphocytic leukemia: Evidence for a multistep pathogenesis of the malignancy. Blood 1991;77:1415–1517. PMID 2009365
- ↑ Bonnet D, Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–737. PMID 9212098
- ↑ Muller-Tidow, C.; Steffen, B.; Cauvet, T.; Tickenbrock, L.; Ji, P.; Diederichs, S.; Sargin, B.; Kohler, G.; Stelljes, M.; Puccetti, E.; Ruthardt, M.; deVos, S.; Hiebert, S. W.; Koeffler, H. P.; Berdel, W. E.; Serve, H. (2004). "Translocation Products in Acute Myeloid Leukemia Activate the Wnt Signaling Pathway in Hematopoietic Cells". Molecular and Cellular Biology. 24 (7): 2890–2904. doi:10.1128/MCB.24.7.2890-2904.2004. ISSN 0270-7306.
- ↑ Abeloff, Martin et al. (2004), pp. 2831–32.
- ↑ Greer, John P., et al. Wintrobe's Clinical Hematology, 11th ed. Philadelphia: Lippincott, Williams, and Wilkins, 2004. p. 2045–2062
- ↑ Melnick A, Licht JD. Deconstructing a disease: RARα its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999;93:3167–3215. PMID 10233871
- ↑ Abeloff, Martin et al. (2004), p. 2828.