Nelfinavir
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Nelfinavir is an antiretroviral that is FDA approved for the treatment of HIV infection. Common adverse reactions include lipodystrophy, diarrhea, fatigue.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
- VIRACEPT in combination with other antiretroviral agents is indicated for the treatment of HIV infection.
Description of Studies
- In the clinical studies described below, efficacy was evaluated by the percent of patients with plasma HIV RNA < 400 copies/mL (Studies 511 and 542) or < 500 copies/mL (Study ACTG 364), using the Roche RT-PCR (Amplicor) HIV-1 Monitor or < 50 copies/mL, using the Roche HIV-1 Ultrasensitive assay (Study Avanti 3). In the analysis presented in each figure, patients who terminated the study early for any reason, switched therapy due to inadequate efficacy or who had a missing HIV-RNA measurement that was either preceded or followed by a measurement above the limit of assay quantification were considered to have HIV-RNA above 400 copies/mL, above 500 copies/mL, or above 50 copies/mL at subsequent time points, depending on the assay that was used.
Studies in Antiretroviral Treatment Naive Patients
- Study 511: VIRACEPT + zidovudine + lamivudine versus zidovudine + lamivudine
- Study 511 was a double-blind, randomized, placebo-controlled trial comparing treatment with zidovudine (ZDV; 200 mg TID) and lamivudine (3TC; 150 mg BID) plus 2 doses of VIRACEPT (750 mg and 500 mg TID) to zidovudine (200 mg TID) and lamivudine (150 mg BID) alone in 297 antiretroviral naive HIV-1 infected patients (median age 35 years [range 21 to 63], 89% male and 78% Caucasian). Mean baseline CD4 cell count was 288 cells/mm3 and mean baseline plasma HIV RNA was 5.21 log10 copies/mL (160,394 copies/mL). The percent of patients with plasma HIV RNA < 400 copies/mL and mean changes in CD4 cell count are summarized in Figures 1 and 2, respectively.
- Figure 1
- Study 511: Percentage of Patients With HIV RNA Below 400 Copies/mL
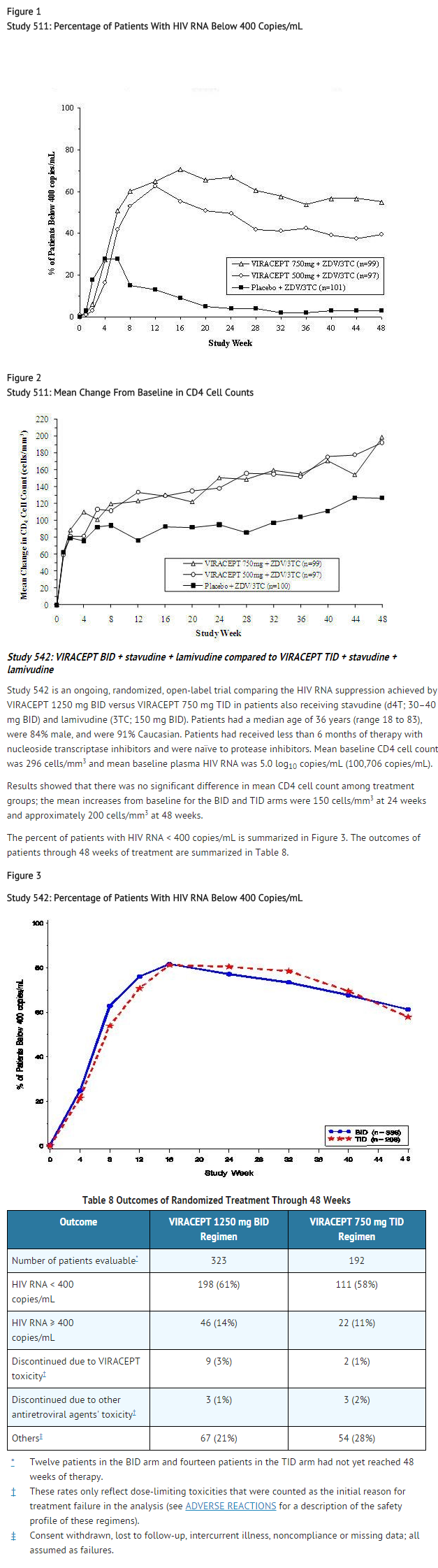
This image is provided by the National Library of Medicine.

- Study 542: VIRACEPT BID + stavudine + lamivudine compared to VIRACEPT TID + stavudine + lamivudine
- Study 542 is an ongoing, randomized, open-label trial comparing the HIV RNA suppression achieved by VIRACEPT 1250 mg BID versus VIRACEPT 750 mg TID in patients also receiving stavudine (d4T; 30–40 mg BID) and lamivudine (3TC; 150 mg BID). Patients had a median age of 36 years (range 18 to 83), were 84% male, and were 91% Caucasian. Patients had received less than 6 months of therapy with nucleoside transcriptase inhibitors and were naïve to protease inhibitors. Mean baseline CD4 cell count was 296 cells/mm3 and mean baseline plasma HIV RNA was 5.0 log10 copies/mL (100,706 copies/mL).
- Results showed that there was no significant difference in mean CD4 cell count among treatment groups; the mean increases from baseline for the BID and TID arms were 150 cells/mm3 at 24 weeks and approximately 200 cells/mm3 at 48 weeks.
- The percent of patients with HIV RNA < 400 copies/mL is summarized in Figure 3. The outcomes of patients through 48 weeks of treatment are summarized in Table 8.
- Figure 3
- Study 542: Percentage of Patients With HIV RNA Below 400 Copies/mL
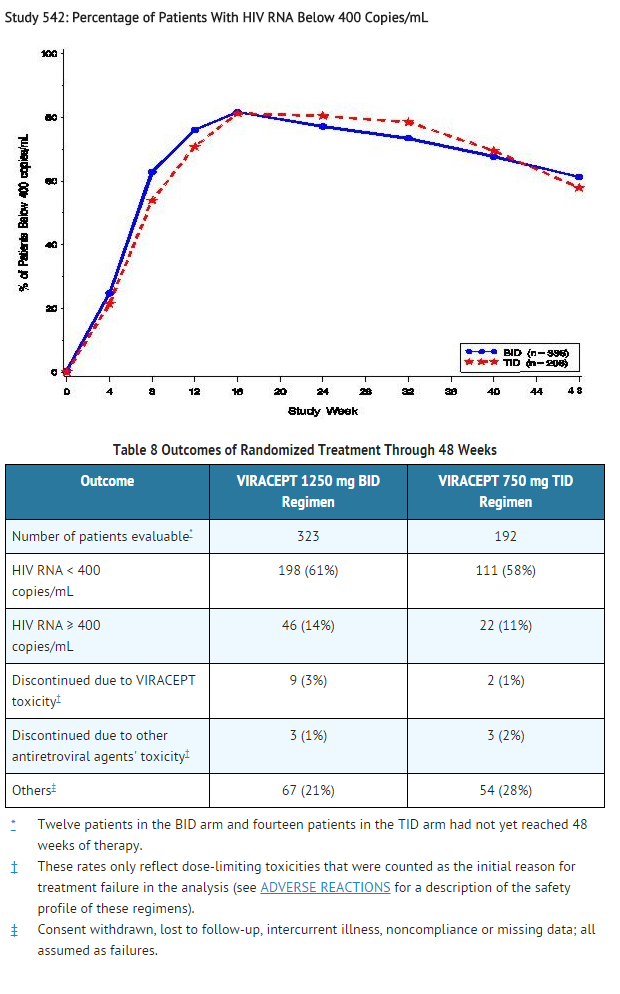
This image is provided by the National Library of Medicine.

- Study Avanti 3: VIRACEPT TID + zidovudine + lamivudine compared to zidovudine + lamivudine
- Study Avanti 3 was a placebo-controlled, randomized, double-blind study designed to evaluate the safety and efficacy of VIRACEPT (750 mg TID) in combination with zidovudine (ZDV; 300 mg BID) and lamivudine (3TC; 150 mg BID) (n=53) versus placebo in combination with ZDV and 3TC (n=52) administered to antiretroviral-naive patients with HIV infection and a CD4 cell count between 150 and 500 cells/µL. Patients had a mean age of 35 (range 22–59), were 89% male, and 88% Caucasian. Mean baseline CD4 cell count was 304 cells/mm3 and mean baseline plasma HIV RNA was 4.8 log10 copies/mL (57,887 copies/mL). The percent of patients with plasma HIV RNA < 50 copies/mL at 52 weeks was 54% for the VIRACEPT + ZDV + 3TC treatment group and 13% for the ZDV + 3TC treatment group.
Studies in Antiretroviral Treatment Experienced Patients
- Study ACTG 364: VIRACEPT TID + 2NRTIs compared to efavirenz + 2NRTIs compared to VIRACEPT + efavirenz + 2NRTIs
- Study ACTG 364 was a randomized, double-blind study that evaluated the combination of VIRACEPT 750 mg TID and/or efavirenz 600 mg QD with 2 NRTIs (either didanosine [ddI] + d4T, ddI + 3TC, or d4T + 3TC) in patients with prolonged prior nucleoside exposure who had completed 2 previous ACTG studies. Patients had a mean age of 41 years (range 18 to 75), were 88% male, and were 74% Caucasian. Mean baseline CD4 cell count was 389 cells/mm3 and mean baseline plasma HIV RNA was 3.9 log10 copies/mL (7,954 copies/mL).
- The percent of patients with plasma HIV RNA < 500 copies/mL at 48 weeks was 42%, 62%, and 72% for the VIRACEPT (n=66), EFV (n=65), and VIRACEPT + EFV (n=64) treatment groups, respectively. The 4-drug combination of VIRACEPT + EFV + 2 NRTIs was more effective in suppressing plasma HIV RNA in these patients than either 3-drug regimen.
Adults
- The recommended dose is 1250 mg (five 250 mg tablets or two 625 mg tablets) twice daily or 750 mg (three 250 mg tablets) three times daily. VIRACEPT should be taken with a meal. Patients unable to swallow the 250 or 625 mg tablets may dissolve the tablets in a small amount of water. Once dissolved, patients should mix the cloudy liquid well, and consume it immediately. The glass should be rinsed with water and the rinse swallowed to ensure the entire dose is consumed.
Pediatric Patients (2–13 years)
- In children 2 years of age and older, the recommended oral dose of VIRACEPT Oral Powder or 250 mg tablets is 45 to 55 mg/kg twice daily or 25 to 35 mg/kg three times daily. All doses should be taken with a meal. Doses higher than the adult maximum dose of 2500 mg per day have not been studied in children. For children unable to take tablets, VIRACEPT Oral Powder may be administered. The oral powder may be mixed with a small amount of water, milk, formula, soy formula, soy milk, or dietary supplements; once mixed, the entire contents must be consumed in order to obtain the full dose. If the mixture is not consumed immediately, it must be stored under refrigeration, but storage must not exceed 6 hours. Acidic food or juice (e.g., orange juice, apple juice, or apple sauce) are not recommended to be used in combination with VIRACEPT, because the combination may result in a bitter taste. VIRACEPT Oral Powder should not be reconstituted with water in its original container.
- The healthcare provider should assess appropriate formulation and dosage for each patient. Crushed 250 mg tablets can be used in lieu of powder. Tables 14 and 15 provide dosing guidelines for VIRACEPT tablets and powder based on age and body weight.
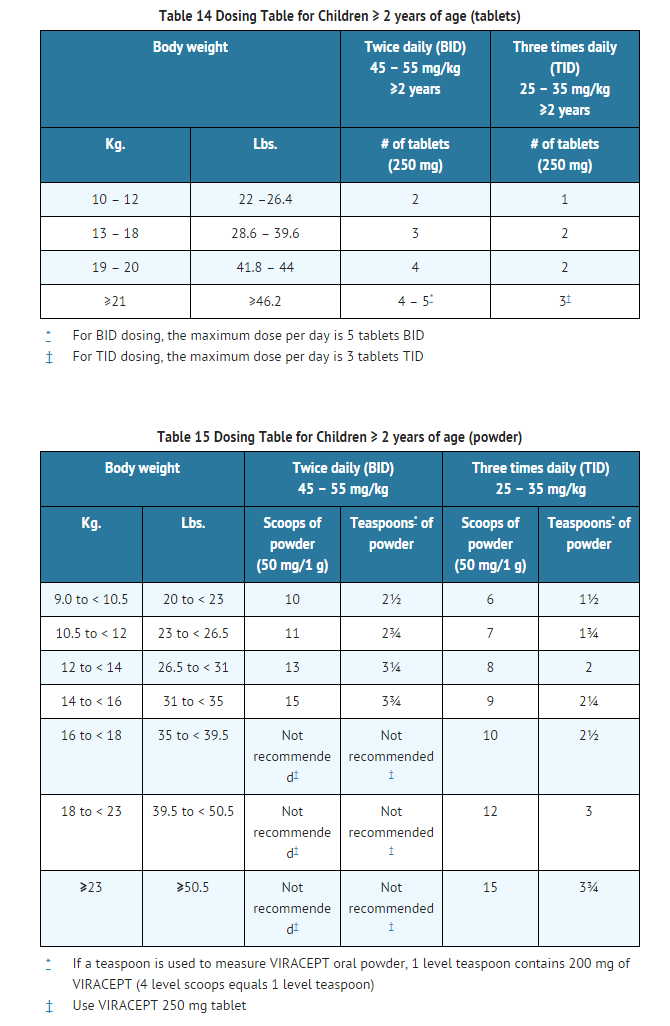
This image is provided by the National Library of Medicine.

Hepatic Impairment
- Viracept can be used in patients with mild hepatic impairment without any dose adjustment. VIRACEPT should not be used in patients with either moderate or severe hepatic impairment.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Nelfinavir in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Nelfinavir in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Nelfinavir in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Nelfinavir in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Nelfinavir in pediatric patients.
Contraindications
- VIRACEPT is contraindicated in patients with clinically significant hypersensitivity to any of its components.
- Coadministration of VIRACEPT is contraindicated with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events. These drugs are listed in Table 9.
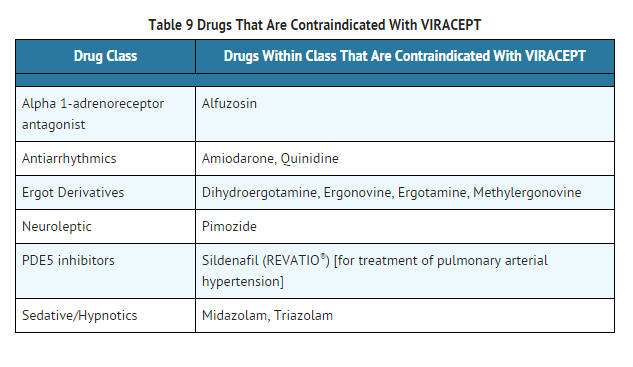
This image is provided by the National Library of Medicine.

Warnings
- ALERT: Find out about medicines that should not be taken with VIRACEPT. This statement is included on the product's bottle label.
Drug Interactions
- Nelfinavir is an inhibitor of the CYP3A enzyme. Coadministration of VIRACEPT and drugs primarily metabolized by CYP3A may result in increased plasma concentrations of the other drug that could increase or prolong its therapeutic and adverse effects. Caution should be exercised when inhibitors of CYP3A, including VIRACEPT, are coadministered with drugs that are metabolized by CYP3A and that prolong the QT interval. Nelfinavir is metabolized by CYP3A and CYP2C19. Coadministration of VIRACEPT and drugs that induce CYP3A or CYP2C19 may decrease nelfinavir plasma concentrations and reduce its therapeutic effect. Coadministration of VIRACEPT and drugs that inhibit CYP3A or CYP2C19 may increase nelfinavir plasma concentrations.
- Concomitant use of VIRACEPT with lovastatin or simvastatin is not recommended. Caution should be exercised if HIV protease inhibitors, including VIRACEPT, are used concurrently with other HMG-CoA reductase inhibitors that are also metabolized by the CYP3A pathway (e.g., atorvastatin). (Also see TABLES 6 and 7: DRUG INTERACTIONS). The risk of myopathy including rhabdomyolysis may be increased when protease inhibitors, including VIRACEPT, are used in combination with these drugs.
- Particular caution should be used when prescribing sildenafil, or other PDE5 inhibitors, in patients receiving protease inhibitors, including VIRACEPT. Coadministration of these drugs is expected to substantially increase PDE5 inhibitor concentrations and may result in an increase in PDE5 inhibitor-associated adverse events, including hypotension, visual changes, and priapism. (See PRECAUTIONS, DRUG INTERACTIONS and INFORMATION FOR PATIENTS, and the complete prescribing information for sildenafil and other PDE5 inhibitors.)
- Concomitant use of St. John's wort (hypericum perforatum) or St. John's wort-containing products and VIRACEPT is not recommended. Coadministration of St. John's wort with protease inhibitors, including VIRACEPT, is expected to substantially decrease protease inhibitor concentrations and may result in sub-optimal levels of VIRACEPT and lead to loss of virologic response and possible resistance to VIRACEPT or to the class of protease inhibitors.
Patients with Phenylketonuria
- Patients with Phenylketonuria: VIRACEPT Oral Powder contains 11.2 mg phenylalanine per gram of powder.
Diabetes mellitus/Hyperglycemia
- New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus and hyperglycemia have been reported during post-marketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
Adverse Reactions
Clinical Trials Experience
- The safety of VIRACEPT was studied in over 5000 patients who received drug either alone or in combination with nucleoside analogues. The majority of adverse events were of mild intensity. The most frequently reported adverse event among patients receiving VIRACEPT was diarrhea, which was generally of mild to moderate intensity.
- Drug-related clinical adverse experiences of moderate or severe intensity in ≥ 2% of patients treated with VIRACEPT coadministered with d4T and 3TC (Study 542) for up to 48 weeks or with ZDV plus 3TC (Study 511) for up to 24 weeks are presented in Table 12.
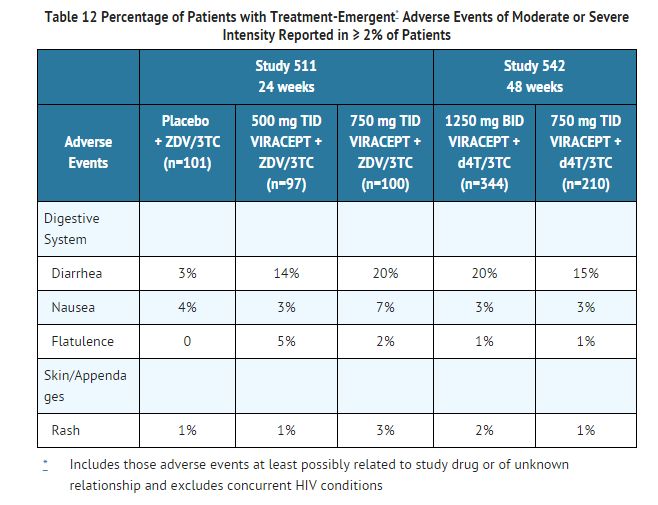
This image is provided by the National Library of Medicine.

- Adverse events occurring in less than 2% of patients receiving VIRACEPT in all phase II/III clinical trials and considered at least possibly related or of unknown relationship to treatment and of at least moderate severity are listed below.
- Body as a Whole: abdominal pain, accidental injury, allergic reaction, asthenia, back pain, fever, headache, malaise, pain, and redistribution/accumulation of body fat.
- Digestive System: anorexia, dyspepsia, epigastric pain, gastrointestinal bleeding, hepatitis, mouth ulceration, pancreatitis, and vomiting.
- Hemic/Lymphatic System: anemia, leukopenia, and thrombocytopenia.
- Metabolic/Nutritional System: increases in alkaline phosphatase, amylase, creatine phosphokinase, lactic dehydrogenase, SGOT, SGPT, and gamma glutamyl transpeptidase; hyperlipemia, hyperuricemia, hyperglycemia, hypoglycemia, dehydration, and liver function tests abnormal.
- Musculoskeletal System: arthralgia, arthritis, cramps, myalgia, myasthenia, and myopathy.
- Nervous System: anxiety, depression, dizziness, emotional lability, hyperkinesia, insomnia, migraine, paresthesia, seizures, sleep disorder, somnolence, and suicide ideation.
- Respiratory System: dyspnea, pharyngitis, rhinitis, and sinusitis.
- Skin/Appendages: dermatitis, folliculitis, fungal dermatitis, maculopapular rash, pruritus, sweating, and urticaria.
- Special Senses: acute iritis and eye disorder.
- Urogenital System: kidney calculus, sexual dysfunction, and urine abnormality.
Postmarketing Experience
- The following additional adverse experiences have been reported from postmarketing surveillance as at least possibly related or of unknown relationship to VIRACEPT:
- Body as a Whole: hypersensitivity reactions (including bronchospasm, moderate to severe rash, fever, and edema).
- Cardiovascular System: QTc prolongation, torsades de pointes.
- Digestive System: jaundice.
- Metabolic/Nutritional System: bilirubinemia, metabolic acidosis.
Laboratory Abnormalities
- The percentage of patients with marked laboratory abnormalities in Studies 542 and 511 are presented in Table 13. Marked laboratory abnormalities are defined as a Grade 3 or 4 abnormality in a patient with a normal baseline value, or a Grade 4 abnormality in a patient with a Grade 1 abnormality at baseline.
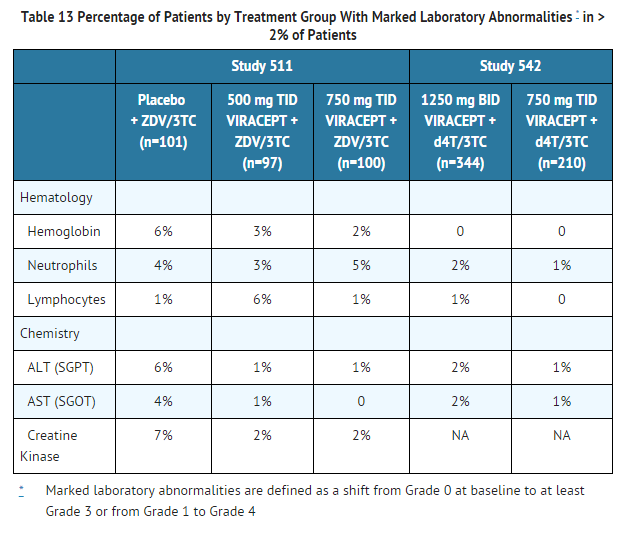
This image is provided by the National Library of Medicine.

Pediatric Population
- VIRACEPT has been studied in approximately 400 pediatric patients in clinical trials from birth to 13 years of age. The adverse event profile seen during pediatric clinical trials was similar to that for adults.
- The most commonly reported drug-related, treatment-emergent adverse events reported in the pediatric studies included: diarrhea, leukopenia/neutropenia, rash, anorexia, and abdominal pain. Diarrhea, regardless of assigned relationship to study drug, was reported in 39% to 47% of pediatric patients receiving VIRACEPT in 2 of the larger treatment trials. Leukopenia/neutropenia was the laboratory abnormality most commonly reported as a significant event across the pediatric studies.
Drug Interactions
There is limited information regarding Nelfinavir Drug Interactions in the drug label.
Use in Specific Populations
Pregnancy
- There were no effects on fetal development or maternal toxicity when nelfinavir was administered to pregnant rats at systemic exposures (AUC) comparable to human exposure. Administration of nelfinavir to pregnant rabbits resulted in no fetal development effects up to a dose at which a slight decrease in maternal body weight was observed; however, even at the highest dose evaluated, systemic exposure in rabbits was significantly lower than human exposure. Additional studies in rats indicated that exposure to nelfinavir in females from mid-pregnancy through lactation had no effect on the survival, growth, and development of the offspring to weaning. Subsequent reproductive performance of these offspring was also not affected by maternal exposure to nelfinavir. However, there are no adequate and well-controlled studies in pregnant women taking VIRACEPT. Because animal reproduction studies are not always predictive of human response, VIRACEPT should be used during pregnancy only if clearly needed.
Antiretroviral Pregnancy Registry: (APR)
- To monitor maternal-fetal outcomes of pregnant women exposed to VIRACEPT and other antiretroviral agents, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling (800) 258-4263.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Nelfinavir in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Nelfinavir during labor and delivery.
Nursing Mothers
- The Centers for Disease Control and Prevention recommends that HIV-infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV. Studies in lactating rats have demonstrated that nelfinavir is excreted in milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breast-feed if they are receiving VIRACEPT.
Pediatric Use
- The safety and effectiveness of VIRACEPT have been established in patients from 2 to 13 years of age. The use of VIRACEPT in these age groups is supported by evidence from adequate and well-controlled studies of VIRACEPT in adults and pharmacokinetic studies and studies supporting activity in pediatric patients. In patients less than 2 years of age, VIRACEPT was found to be safe at the doses studied, but a reliably effective dose could not be established.
- The following issues should be considered when initiating VIRACEPT in pediatric patients:
- In pediatric patients ≥ 2 years of age receiving VIRACEPT as part of triple combination antiretroviral therapy in randomized studies, the proportion of patients achieving a HIV RNA level <400 copies/mL through 48 weeks ranged from 26% to 42%.
- Response rates in children <2 years of age appeared to be poorer than those in patients ≥ 2 years of age in some studies.
- Highly variable drug exposure remains a significant problem in the use of VIRACEPT in pediatric patients. Unpredictable drug exposure may be exacerbated in pediatric patients because of increased clearance compared to adults and difficulties with compliance and adequate food intake with dosing. Pharmacokinetic results from the pediatric studies are reported in Table 5.
- Study 556 was a randomized, double-blind, placebo-controlled trial with VIRACEPT or placebo coadministered with ZDV and ddI in 141 HIV-positive children who had received minimal antiretroviral therapy. The mean age of the children was 3.9 years. Ninety four (67%) children were between 2–12 years, and 47 (33%) were < 2 years of age. The mean baseline HIV RNA value was 5.0 log for all patients and the mean CD4 cell count was 886 cells/mm3 for all patients. The efficacy of VIRACEPT measured by HIV RNA <400 at 48 weeks in children ≥ 2 years of age was 26% compared to 2% of placebo patients (p=0.0008). In the children < 2 years of age, only 1 of 27 and 2 of 20 maintained an undetectable HIV RNA level at 48 weeks for placebo and VIRACEPT patients, respectively.
- PACTG 377 was an open-label study that randomized 181 HIV treatment-experienced pediatric patients to receive: d4T+NVP+RTV, d4T+3TC+NFV, or d4T+3TC+NVP+NFV with NFV given on a TID schedule. The median age was 5.9 years and 46% were male. At baseline the median HIV RNA was 4.4 log and median CD4 cell count was 690 cells/mm3. Substudy PACTG 725 evaluated d4T+3TC+NFV with NFV given on a BID schedule. The proportion of patients with detectable viral load at baseline achieving HIV RNA <400 copies/mL at 48 weeks was: 41% for d4T+NVP+RTV, 42% for d4T+3TC+NFV, 30% for d4T+NVP+NFV, and 52% for d4T+3TC+NVP+NFV. No significant clinical differences were identified between patients receiving VIRACEPT in BID or TID schedules.
- VIRACEPT has been evaluated in 2 studies of young infants. The PENTA 7 study was an open-label study to evaluate the toxicity, tolerability, pharmacokinetics, and activity of NFV+d4T+ddI in 20 HIV-infected infants less than 12 weeks of age. PACTG 353 evaluated the pharmacokinetics and safety of VIRACEPT in infants born to HIV-infected women receiving NFV as part of combination therapy during pregnancy.
Geriatic Use
- Clinical studies of VIRACEPT did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
Gender
There is no FDA guidance on the use of Nelfinavir with respect to specific gender populations.
Race
There is no FDA guidance on the use of Nelfinavir with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Nelfinavir in patients with renal impairment.
Hepatic Impairment
There is no FDA guidance on the use of Nelfinavir in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Nelfinavir in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Nelfinavir in patients who are immunocompromised.
Administration and Monitoring
Administration
Monitoring
There is limited information regarding Monitoring of Nelfinavir in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Nelfinavir in the drug label.
Overdosage
- Human experience of acute overdose with VIRACEPT is limited. There is no specific antidote for overdose with VIRACEPT. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage. Administration of activated charcoal may also be used to aid removal of unabsorbed drug. Since nelfinavir is highly protein bound, dialysis is unlikely to significantly remove drug from blood.
Pharmacology
There is limited information regarding Nelfinavir Pharmacology in the drug label.
Mechanism of Action
Structure
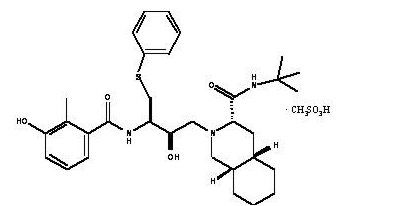
This image is provided by the National Library of Medicine.

Pharmacodynamics
There is limited information regarding Pharmacodynamics of Nelfinavir in the drug label.
Pharmacokinetics
- The pharmacokinetic properties of nelfinavir were evaluated in healthy volunteers and HIV-infected patients; no substantial differences were observed between the two groups.
Absorption
- Pharmacokinetic parameters of nelfinavir (area under the plasma concentration-time curve during a 24-hour period at steady-state [AUC24], peak plasma concentrations [Cmax], morning and evening trough concentrations [Ctrough]) from a pharmacokinetic study in HIV-positive patients after multiple dosing with 1250 mg (five 250 mg tablets) twice daily (BID) for 28 days (10 patients) and 750 mg (three 250 mg tablets) three times daily (TID) for 28 days (11 patients) are summarized in Table 1.
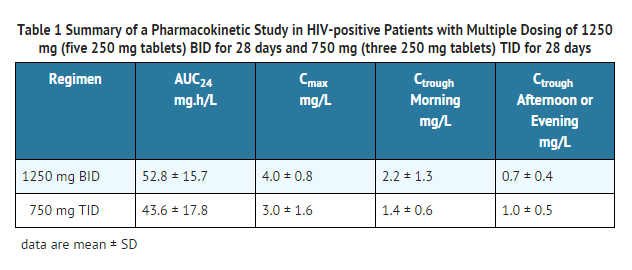
This image is provided by the National Library of Medicine.

- The difference between morning and afternoon or evening trough concentrations for the TID and BID regimens was also observed in healthy volunteers who were dosed at precisely 8- or 12-hour intervals.
- In healthy volunteers receiving a single 1250 mg dose, the 625 mg tablet was not bioequivalent to the 250 mg tablet formulation. Under fasted conditions (n=27), the AUC and Cmax were 34% and 24% higher, respectively, for the 625 mg tablets. In a relative bioavailability assessment under fed conditions (n=28), the AUC was 24% higher for the 625 mg tablet; the Cmax was comparable for both formulations. In HIV-1 infected subjects (N = 21) receiving multiple doses of 1250 mg BID under fed conditions, the 625 mg formulation was bioequivalent to the 250 mg formulation based on similarity in steady state exposure (Cmax and AUC).
- Table 2 shows the summary of the steady state pharmacokinetic parameters (mean ± SD) of nelfinavir after multiple dose administration of 1250 mg BID (2 × 625 tablets) to HIV-infected patients (N = 21) for 14 days.
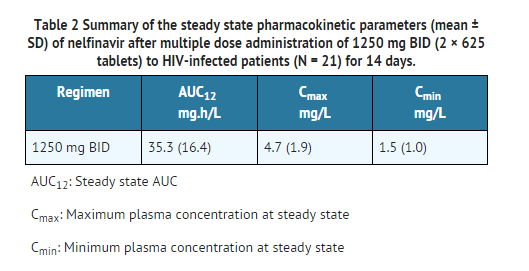
This image is provided by the National Library of Medicine.

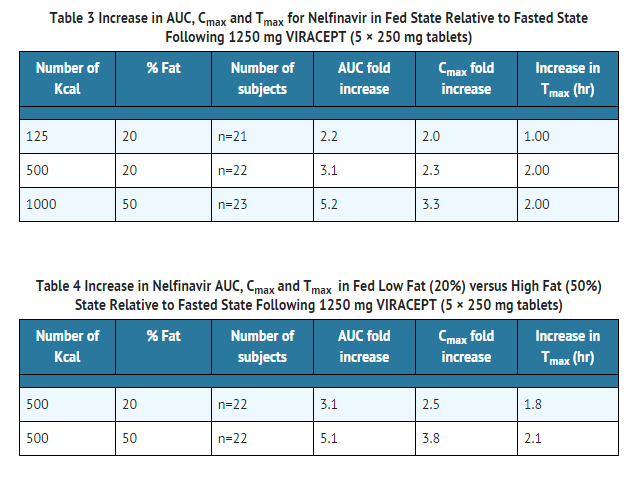
Nelfinavir exposure can be increased by increasing the calorie or fat content in meals taken with VIRACEPT.
- A food effect study has not been conducted with the 625 mg tablet. However, based on a cross-study comparison (n=26 fed vs. n=26 fasted) following single dose administration of nelfinavir 1250 mg, the magnitude of the food effect for the 625 mg nelfinavir tablet appears comparable to that of the 250 mg tablets. VIRACEPT should be taken with a meal.
Distribution
- The apparent volume of distribution following oral administration of nelfinavir was 2–7 L/kg. Nelfinavir in serum is extensively protein-bound (>98%).
Metabolism
- Unchanged nelfinavir comprised 82–86% of the total plasma radioactivity after a single oral 750 mg dose of 14C-nelfinavir. In vitro, multiple cytochrome P-450 enzymes including CYP3A and CYP2C19 are responsible for metabolism of nelfinavir. One major and several minor oxidative metabolites were found in plasma. The major oxidative metabolite has in vitro antiviral activity comparable to the parent drug.
Elimination
- The terminal half-life in plasma was typically 3.5 to 5 hours. The majority (87%) of an oral 750 mg dose containing 14C-nelfinavir was recovered in the feces; fecal radioactivity consisted of numerous oxidative metabolites (78%) and unchanged nelfinavir (22%). Only 1–2% of the dose was recovered in urine, of which unchanged nelfinavir was the major component.
Special Populations
Hepatic Insufficiency
- The steady-state pharmacokinetics of nelfinavir (1250 mg BID for 2 weeks) was studied in HIV-seronegative subjects with mild (Child-Pugh Class A; n=6) or moderate (Child-Pugh Class B; n=6) hepatic impairment. When compared with subjects with normal hepatic function, the Cmax and AUC of nelfinavir were not significantly different in subjects with mild hepatic impairment but were increased by 22% and 62%, respectively, in subjects with moderate hepatic impairment. The steady-state pharmacokinetics of nelfinavir has not been studied in HIV-seronegative subjects with severe hepatic impairment.
- The steady-state pharmacokinetics of nelfinavir has not been studied in HIV-positive patients with any degree of hepatic impairment.
Renal Insufficiency
- The pharmacokinetics of nelfinavir have not been studied in patients with renal insufficiency; however, less than 2% of nelfinavir is excreted in the urine, so the impact of renal impairment on nelfinavir elimination should be minimal.
Gender and Race
- No significant pharmacokinetic differences have been detected between males and females. Pharmacokinetic differences due to race have not been evaluated.
Pediatrics
- The pharmacokinetics of nelfinavir have been investigated in 5 studies in pediatric patients from birth to 13 years of age either receiving VIRACEPT three times or twice daily. The dosing regimens and associated AUC24 values are summarized in Table 5.
This image is provided by the National Library of Medicine.

Nonclinical Toxicology
There is limited information regarding Nonclinical Toxicology of Nelfinavir in the drug label.
Clinical Studies
There is limited information regarding Nelfinavir Clinical Studies in the drug label.
How Supplied
- VIRACEPT (nelfinavir mesylate) 250 mg: Light blue, capsule-shaped tablets with a clear film coating engraved with "VIRACEPT" on one side and "250 mg" on the other.
- Bottles of 300, 250 mg tablets………………………………...NDC 63010-010-30
- VIRACEPT (nelfinavir mesylate) 625 mg: White oval tablet with a clear film coating engraved with "V" on one side and "625" on the other.
- Bottles of 120, 625 mg tablets………………………………...NDC 63010-027-70
- VIRACEPT (nelfinavir mesylate) Oral Powder is available as a 50 mg/g off-white powder containing 50 mg (as nelfinavir free base) in each level scoopful (1 gram).
- Multiple use bottles of 144 grams of powder with scoop …….NDC 63010-011-90
Storage
- Viracept tablets and oral powder should be stored at 15° to 30°C (59° TO 86°F).
- Keep container tightly closed. Dispense in original container.
Images
Drug Images
{{#ask: Page Name::Nelfinavir |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Nelfinavir |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
There is limited information regarding Nelfinavir Patient Counseling Information in the drug label.
Precautions with Alcohol
- Alcohol-Nelfinavir interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- VIRACEPT ®[1]
Look-Alike Drug Names
There is limited information regarding Nelfinavir Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Label Page=Nelfinavir |Label Name=Nelvi 15 Ingredients.png
}}
{{#subobject:
|Label Page=Nelfinavir |Label Name=Nelvi 15 pack.jpg
}}