Gaucher's disease: Difference between revisions
| Line 148: | Line 148: | ||
[[Image:Gaucher disease 007.jpg|left|thumb|300px|]] | [[Image:Gaucher disease 007.jpg|left|thumb|300px|]] | ||
<br clear="left"/> | |||
[[Image:Gaucher disease 008.jpg|left|thumb|300px|]] | |||
<br clear="left"/> | <br clear="left"/> | ||
Revision as of 04:29, 22 February 2009
| Gaucher's disease | |
 | |
|---|---|
| MRI: H-shaped vertebral bodies in a patient with Gaucher's disease. (Image courtesy of RadsWiki) | |
| ICD-10 | E75.2 (ILDS E75.220) |
| ICD-9 | 272.7 |
| OMIM | 230800 230900 231000 |
| DiseasesDB | 5124 |
| MedlinePlus | 000564 |
| eMedicine | ped/837 derm/709 |
| MeSH | D005776 |
|
WikiDoc Resources for Gaucher's disease |
|
Articles |
|---|
|
Most recent articles on Gaucher's disease Most cited articles on Gaucher's disease |
|
Media |
|
Powerpoint slides on Gaucher's disease |
|
Evidence Based Medicine |
|
Cochrane Collaboration on Gaucher's disease |
|
Clinical Trials |
|
Ongoing Trials on Gaucher's disease at Clinical Trials.gov Trial results on Gaucher's disease Clinical Trials on Gaucher's disease at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Gaucher's disease NICE Guidance on Gaucher's disease
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Gaucher's disease Discussion groups on Gaucher's disease Patient Handouts on Gaucher's disease Directions to Hospitals Treating Gaucher's disease Risk calculators and risk factors for Gaucher's disease
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Gaucher's disease |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Please Take Over This Page and Apply to be Editor-In-Chief for this topic: There can be one or more than one Editor-In-Chief. You may also apply to be an Associate Editor-In-Chief of one of the subtopics below. Please mail us [2] to indicate your interest in serving either as an Editor-In-Chief of the entire topic or as an Associate Editor-In-Chief for a subtopic. Please be sure to attach your CV and or biographical sketch.
Overview
Gaucher's disease (pronounced {{goshay}}) is the most common of the lysosomal storage diseases. It is caused by a deficiency of the enzyme glucocerebrosidase, leading to an accumulation of its substrate, the fatty substance glucocerebroside (also known as glucosylceramide). Fatty material can collect in the spleen, liver, kidneys, lungs, brain and bone marrow. Symptoms may include enlarged spleen and liver, liver malfunction, skeletal disorders and bone lesions that may cause pain, severe neurologic complications, swelling of lymph nodes and (occasionally) adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelets and yellow fatty deposits on the sclera. Persons affected most seriously may also be more susceptible to infection.
Genetics
The disease shows autosomal recessive inheritance, therefore affects males and females equally.
Etymology
It is named after the French doctor Philippe Gaucher who originally described it in 1882.
History
Philippe Gaucher described the disease in his doctoral thesis in 1882.[1] The biochemical basis for the disease would be elucidated in 1965 by Brady et al.[2]
Epidemiology
- The National Gaucher Foundation states that around 1 in 100 people in the general U.S. population is a carrier for type 1 Gaucher's disease, giving a prevalence of 1 in 40000: the rate of carriers is considerably higher, at roughly 1 in 15, among Ashkenazi Jews.[3]
- Type 2 Gaucher's disease shows no particular preference for any ethnic group.
- Type 3 Gaucher's disease is especially common in the population of the Northern Swedish region of Norrbotten where the incidence of the disease is 1 in 50.000.
Pathophysiology

The disease is caused by a defect in the housekeeping gene lysosomal gluco-cerebrosidase (also known as β-glucosidase, EC 3.2.1.45, PDB: 1OGS) on the first chromosome (1q21).
The enzyme is a 55.6 KD, 497 amino acids long protein that catalyses the breakdown of glucocerebroside, a cell membrane constituent of red and white blood cells.
The macrophages that clear these cells are unable to eliminate the waste product, which accumulates in fibrils, and turn into Gaucher cells, which appear on light microscopy as appearing to contain crumpled-up paper.
Different mutations in the β-glucosidase determine the remaining activity of the enzyme, and, to a large extent, the phenotype.
In the brain (type II and III), glucocerebroside accumulates due to the turnover of complex lipids during brain development and the formation of the myelin sheath of nerves.
Research suggests that heterozygotes for particular acid β-glucosidase mutations are at an increased risk of Parkinson's disease.[4]
A study of 1525 Gaucher patients in the United States suggested that while cancer risk is not elevated, particular malignancies (non-Hodgkin lymphoma, melanoma and pancreatic cancer) occurred at a 2-3 times higher rate.[5]
Classification and genetics
The three types of Gaucher's disease are inherited in an autosomal recessive fashion. Both parents must be carriers in order for a child to be affected. If both parents are carriers, there is a one in four, or 25%, chance with each pregnancy for an affected child. Genetic counseling and genetic testing is recommended for families who may be carriers of mutations.
Each type has been linked to particular mutations. In all, there are about 80 known mutations, grouped into three main types:[6]
- Type I (N370S homozygote, the most common, also called the "non-neuropathic" type) occurs mainly (100x the general populace) in Ashkenazi Jews. It is mainly diagnosed in late childhood or early adulthood. Life expectancy is mildly decreased. There are no neurological symptoms. Dor Yeshorim, a non-profit testing organisation, therefore only tests patients on request.
- Type II (1 or 2 alleles L444P) is characterized by neurological problems in small children. The enzyme is hardly released into the lysosomes. Prognosis is dismal: most die before reaching the third birthday.
- Type III (also 1-2 copies of L444P, possibly delayed by protective polymorphisms) occurs in Swedish patients from the Norrbotten region. This group develops the disease somewhat later, but most die before their 30th birthday.
Diaz et al suggest that the Gaucher-causing mutations entered the Ashkenazi Jewish gene pool in the early Middle Ages (48-55 generations ago).[7]
Subtypes
Gaucher's disease has three common clinical subtypes.
- Type I (or nonneuropathic type) is the most common form of the disease, occurring in approximately 1 in 50,000 live births. It occurs most often among persons of Ashkenazi Jewish heritage. Symptoms may begin early in life or in adulthood and include enlarged liver and grossly enlarged spleen, which can rupture and cause additional complications. Skeletal weakness and bone disease may be extensive. Spleen enlargement and bone marrow replacement cause anemia, thrombocytopenia and leukopenia. The brain is not affected, but there may be lung and, rarely, kidney impairment. Patients in this group usually bruise easily and experience fatigue due to low blood platelets. Depending on disease onset and severity, type 1 patients may live well into adulthood. Many patients have a mild form of the disease or may not show any symptoms.
- Type II (or acute infantile neuropathic Gaucher's disease) typically begins within 6 months of birth and has an incidence rate of approximately 1 in 100,000 live births. Symptoms include an enlarged liver and spleen, extensive and progressive brain damage, eye movement disorders, spasticity, seizures, limb rigidity, and a poor ability to suck and swallow. Affected children usually die by age 2.
- Type III (the chronic neuronopathic form) can begin at any time in childhood or even in adulthood, and occurs in approximately 1 in 100,000 live births. It is characterized by slowly progressive but milder neurologic symptoms compared to the acute or type 2 version. Major symptoms include an enlarged spleen and/or liver, seizures, poor coordination, skeletal irregularities, eye movement disorders, blood disorders including anemia and respiratory problems. Patients often live into their early teen years and adulthood.
These subtypes have come under some criticism for not taking account of the spectrum of phenotypes.[3] There are also compound heterozygous variations which considerably increase the complexity of predicting disease course.
Signs and symptoms
- Painless hepatomegaly and splenomegaly; the spleen can be 1500-3000 ml, as opposed to the normal size of 50-200 ml.
- Hypersplenism: increased destruction of red and white blood cells and platelets, leading to anemia, neutropenia and thrombocytopenia (with an increased risk of infection and bleeding)
- Cirrhosis of the liver is rare
- Neurological symptoms occur only in some types of Gaucher's (see below):
- Type II: serious convulsions, hypertonia, mental retardation, apnea.
- Type III: myoclonus, convulsions, dementia, ocular muscle apraxia.
- Osteoporosis: 75% develop visible bony abnormalities due to the accumulated glucosylceramide. An Erlenmeyer flask deformity of the distal femur is commonly described
- Yellowish-brown skin pigmentation
- No cardiac, renal and pulmonary signs
Diagnosis
In populations with high rates of carriage (Ashkenazi Jews and Norrbottnian Swedes and a few African American tribes), some family members of the index patient may already have been diagnosed with Gaucher's. Truly sporadic cases may suffer diagnostic delay due to the protean symptoms.
Biochemical abnormalities: high alkaline phosphatase, angiotensin-converting enzyme (ACE) and immunoglobulin levels.
The diagnosis is made with genetic testing of the β-glucosidase gene. As there are numerous different mutations, sequencing of the gene is sometimes necessary to confirm the diagnosis. Prenatal diagnosis is available, and is useful when there is a known genetic risk factor.
Diagnostic Findings
MR images demonstrate Erlenmeyer flask deformities and H-shaped vertebral bodies in a patient with Gaucher's disease
(Images courtesy of RadsWiki)
-
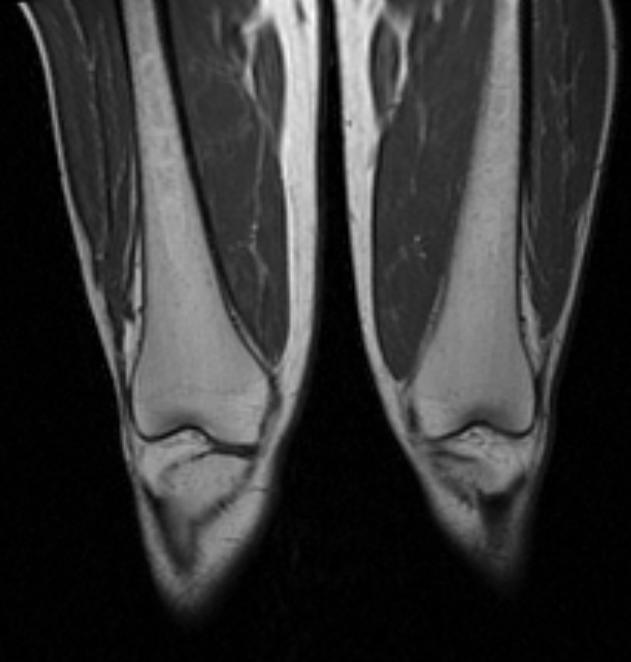
Erlenmeyer flask deformity in a patient with Gaucher's disease
-
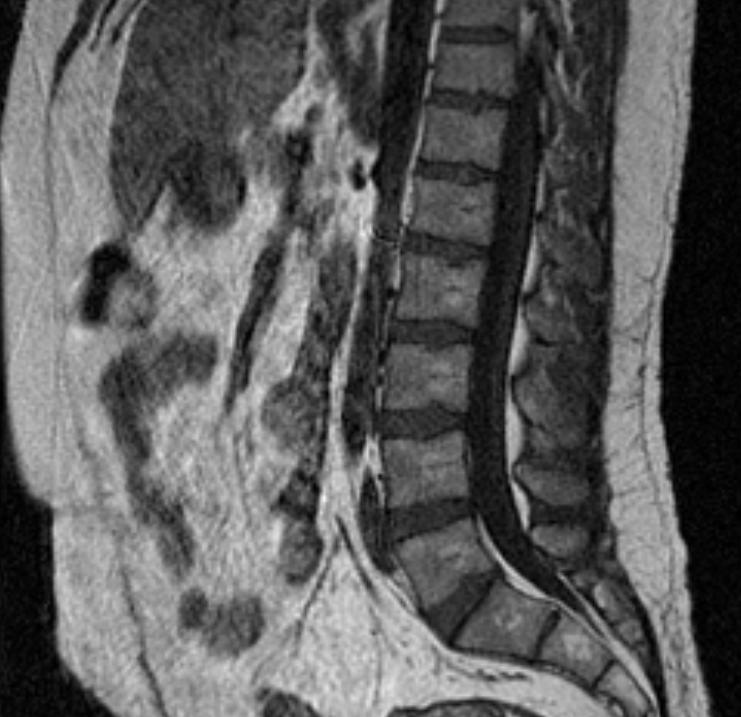
H-shaped vertebral bodies in a patient with Gaucher's disease

Treatment
For type 1 and most type 3 patients, enzyme replacement treatment with mannose-terminated recombinant glucocerebrosidase, 60 Units/kg, given intravenously every two weeks can dramatically decrease liver and spleen size, reduce skeletal abnormalities, and reverse other manifestations. This treatment is becoming the standard in treating Gaucher's. Due to the low incidence, this has become an orphan drug in many countries. Successful bone marrow transplantation cures the non-neurological manifestations of the disease, because it introduces a monocyte population with active β-glucosidase. However, this procedure carries significant risk and is rarely performed in Gaucher patients. Surgery to remove the spleen (splenectomy) may be required on rare occasions if the patient is anemic or when the enlarged organ affects the patient’s comfort. Blood transfusion may benefit some anemic patients. Other patients may require joint replacement surgery to improve mobility and quality of life. Other treatment options include antibiotics for infections, antiepileptics for seizures, bisphosphonates for bone lesions, and liver transplants. Substrate reduction therapy may prove to be effective in stopping Type 2, as it can cross through the blood barrier into the brain. There is currently no effective treatment for the severe brain damage that may occur in patients with types 2 and 3 Gaucher disease. Gene therapy may be a future step.
Gaucher's disease has recently become a target for more than one effort at pharmacological chaperoning since the crystal structure of glucocerebrosidase is known.
The currently existing treatment of Gaucher's disease, Cerezyme (imiglucerase for injection), costs up to $550,000 annually for a single patient and the treatment should be continued for life. This recombinant β-glucosidase is given intravenously. Miglustat is another drug approved for this disease in 2003.
Case Examples
Case #1
Clinical Summary
A 23-year-old black female delivered a stillborn infant by Caesarean section, following which she experienced excessive uterine bleeding including the passage of blood clots. Further study revealed an enlarged spleen and thrombocytopenia (platelet count 58,000). A splenectomy was performed with an uneventful postoperative course other than persistent pain in her right leg (the right hip had been fractured 2 years earlier). Subsequently she developed aseptic necrosis of the left femoral head requiring a prosthetic replacement. Microscopic evaluation of the bone removed at the time of surgery revealed Gaucher cells in the marrow space.
Autopsy Findings
The surgical specimen was a 935-gram spleen. Its surface was pale with an area of bluish discoloration. The cut surface revealed the same pale appearance.
Histopathological Findings








References
- ↑ Gaucher PCE. De l'epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie. Academic thesis, Paris, France, 1882.
- ↑ Brady RO, Kanfer JN, Shapiro D (1965). "Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher's disease". Biochem. Biophys. Res. Commun. 18: 221–5. PMID 14282020.
- ↑ "National Gaucher Foundation". Retrieved 2007-05-30.
- ↑ Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004). "Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews". N. Engl. J. Med. 351 (19): 1972–7. doi:10.1056/NEJMoa033277. PMID 15525722.
- ↑ Landgren O, Turesson I, Gridley G, Caporaso NE (2007). "Risk of Malignant Disease Among 1525 Adult Male US Veterans With Gaucher Disease". 167 (11): 1189–1194. doi:10.1001/archinte.167.11.1189. PMID 17563029.
- ↑ Online Mendelian Inheritance in Man (OMIM) 606463
- ↑ Diaz GA, Gelb BD, Risch N; et al. (2000). "Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid beta-glucosidase mutations". Am. J. Hum. Genet. 66 (6): 1821–32. PMID 10777718.
External links
Template:Endocrine, nutritional and metabolic pathology Template:SIB de:Morbus Gaucher he:מחלת גושה