Cardiomyopathy pathophysiology: Difference between revisions
| Line 3: | Line 3: | ||
{{CMG}} | {{CMG}} | ||
==Overview== | ==Overview== | ||
Different causes of cardiomyopathies, including genetic and acquired causes, result in abnormal heart structure and function. As the function of the heart deteriorates, symptoms of heart failure become apparent. On the other hand, defects in ion channels and hypertrophic cardiomyopathy can present with fatal arrhythmias and sudden cardiac death without the preceding symptoms of heart failure. In DCM, the heart (especially the [[left ventricle]]) is enlarged and the pumping function is diminished. Approximately 40% of cases are familial. Mutations in | Different causes of cardiomyopathies, including genetic and acquired causes, result in abnormal heart structure and function. As the function of the heart deteriorates, symptoms of heart failure become apparent. On the other hand, defects in ion channels and hypertrophic cardiomyopathy can present with fatal arrhythmias and sudden cardiac death without the preceding symptoms of heart failure. In DCM, the heart (especially the [[left ventricle]]) is enlarged and the pumping function is diminished. Approximately 40% of cases are familial. Mutations in multiple genes have been described. In some cases it manifests as [[peripartum cardiomyopathy]], and in other cases it may be associated with alcoholism.[[Hypertrophic cardiomyopathy]](HCM or HOCM), a [[genetic disorder]] caused by various [[mutation]][[mutation|<nowiki/>]]s in genes encoding [[Sarcomere|sarcomer]][[Sarcomere|ic]] proteins. In HCM the heart muscle is thickened, which can obstruct blood flow and prevent the heart from functioning properly. [[Arrhythmogenic right ventricular cardiomyopathy]](ARVC) arises from an electrical disturbance of the heart in which heart muscle is replaced by fibrous scar tissue. The [[right ventricle]] is generally most affected, but LV can also be affected. [[Restrictive cardiomyopathy]](RCM) is an uncommon cardiomyopathy. The walls of the ventricles are stiff, but may not be thickened, and resist the normal filling of the heart with blood. A rare form of restrictive cardiomyopathy is the obliterative cardiomyopathy in [[hypereosinophilic syndrome]]. In this type, the myocardium in the apices of the left and right ventricles becomes thickened and fibrotic, causing a decrease in ventricular volumes. [[Noncompaction cardiomyopathy]] has been recognized as a separate type since the 1980s. The term refers to a cardiomyopathy where the left ventricle wall has failed to grow properly from birth and has a spongy appearance on an echocardiogram. These patients are at risk of heart failure, thromboembolic events, and sudden cardiac death. | ||
<nowiki/><nowiki/><nowiki/>[[mutation|<nowiki/>]] | |||
==Cardiomyopathy Pathophysiology== | ==Cardiomyopathy Pathophysiology== | ||
Different causes of cardiomyopathies, including genetic and acquired causes, result in abnormal heart structure and function. As the function of the heart deteriorates, symptoms of heart failure become apparent. On the other hand, defects in ion channels and hypertrophic cardiomyopathy can present with fatal arrhythmias and sudden cardiac death without the preceding symptoms of heart failure. | Different causes of cardiomyopathies, including genetic and acquired causes, result in abnormal heart structure and function. As the function of the heart deteriorates, symptoms of heart failure become apparent. On the other hand, defects in ion channels and hypertrophic cardiomyopathy can present with fatal arrhythmias and sudden cardiac death without the preceding symptoms of heart failure. | ||
*In DCM, the heart (especially the [[left ventricle]]) is enlarged and the pumping function is diminished. Approximately 40% of cases are familial. Mutations in mu<nowiki/>ltiple genes have been described. In some cases it manifests as [[peripartum cardiomyopathy]], and in other cases it may be associated with alcoholism. | *In DCM, the heart (especially the [[left ventricle]]) is enlarged and the pumping function is diminished. Approximately 40% of cases are familial. Mutations in mu<nowiki/>ltiple genes have been described. In some cases it manifests as [[peripartum cardiomyopathy]], and in other cases it may be associated with alcoholism. | ||
| Line 11: | Line 13: | ||
*[[Arrhythmogenic right ventricular cardiomyopathy]](ARVC) arises from an electrical disturbance of the heart in which heart muscle is replaced by fibrous scar tissue. The [[right ventricle]]<nowiki/> is generally most affected, but LV can also be affected. | *[[Arrhythmogenic right ventricular cardiomyopathy]](ARVC) arises from an electrical disturbance of the heart in which heart muscle is replaced by fibrous scar tissue. The [[right ventricle]]<nowiki/> is generally most affected, but LV can also be affected. | ||
*[[Restrictive cardiomyopathy]](RCM) is an uncommon cardiomyopathy. The walls of the ventricles are stiff, but may not be thickened, and resist the normal filling of the heart with blood. A rare form of restrictive cardiomyopathy is the obliterative cardiomyopathy in [[hypereosinophilic syndrome]]. In this type, the myocardium in the apices of the left and right ventricles becomes thickened and fibrotic, causing a decrease in ventricular volumes. | *[[Restrictive cardiomyopathy]](RCM) is an uncommon cardiomyopathy. The walls of the ventricles are stiff, but may not be thickened, and resist the normal filling of the heart with blood. A rare form of restrictive cardiomyopathy is the obliterative cardiomyopathy in [[hypereosinophilic syndrome]]. In this type, the myocardium in the apices of the left and right ventricles becomes thickened and fibrotic, causing a decrease in ventricular volumes. | ||
*[[Noncompaction cardiomyopathy]]<nowiki/>has been recognized as a separate type since the 1980s. The term refers to a cardiomyopathy where the left ventricle wall has failed to grow properly from birth and has a spongy appearance on echocardiogram. These patients are at risk of heart failure, thromboembolic events and sudden cardiac death | *[[Noncompaction cardiomyopathy]]<nowiki/> has been recognized as a separate type since the 1980s. The term refers to a cardiomyopathy where the left ventricle wall has failed to grow properly from birth and has a spongy appearance on echocardiogram. These patients are at risk of heart failure, thromboembolic events and sudden cardiac death | ||
===Gross Pathology=== | ===Gross Pathology=== | ||
[http://www.peir.net Images shown below are courtesy of Professor Peter Anderson DVM PhD and published with permission © PEIR, University of Alabama at Birmingham, Department of Pathology] | [http://www.peir.net Images shown below are courtesy of Professor Peter Anderson DVM PhD and published with permission © PEIR, University of Alabama at Birmingham, Department of Pathology] | ||
Revision as of 17:51, 3 February 2019
|
Cardiomyopathy Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Guidelines |
|
2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy |
|
Case Studies |
|
Cardiomyopathy pathophysiology On the Web |
|
American Roentgen Ray Society Images of Cardiomyopathy pathophysiology |
|
Risk calculators and risk factors for Cardiomyopathy pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
Different causes of cardiomyopathies, including genetic and acquired causes, result in abnormal heart structure and function. As the function of the heart deteriorates, symptoms of heart failure become apparent. On the other hand, defects in ion channels and hypertrophic cardiomyopathy can present with fatal arrhythmias and sudden cardiac death without the preceding symptoms of heart failure. In DCM, the heart (especially the left ventricle) is enlarged and the pumping function is diminished. Approximately 40% of cases are familial. Mutations in multiple genes have been described. In some cases it manifests as peripartum cardiomyopathy, and in other cases it may be associated with alcoholism.Hypertrophic cardiomyopathy(HCM or HOCM), a genetic disorder caused by various mutations in genes encoding sarcomeric proteins. In HCM the heart muscle is thickened, which can obstruct blood flow and prevent the heart from functioning properly. Arrhythmogenic right ventricular cardiomyopathy(ARVC) arises from an electrical disturbance of the heart in which heart muscle is replaced by fibrous scar tissue. The right ventricle is generally most affected, but LV can also be affected. Restrictive cardiomyopathy(RCM) is an uncommon cardiomyopathy. The walls of the ventricles are stiff, but may not be thickened, and resist the normal filling of the heart with blood. A rare form of restrictive cardiomyopathy is the obliterative cardiomyopathy in hypereosinophilic syndrome. In this type, the myocardium in the apices of the left and right ventricles becomes thickened and fibrotic, causing a decrease in ventricular volumes. Noncompaction cardiomyopathy has been recognized as a separate type since the 1980s. The term refers to a cardiomyopathy where the left ventricle wall has failed to grow properly from birth and has a spongy appearance on an echocardiogram. These patients are at risk of heart failure, thromboembolic events, and sudden cardiac death.
Cardiomyopathy Pathophysiology
Different causes of cardiomyopathies, including genetic and acquired causes, result in abnormal heart structure and function. As the function of the heart deteriorates, symptoms of heart failure become apparent. On the other hand, defects in ion channels and hypertrophic cardiomyopathy can present with fatal arrhythmias and sudden cardiac death without the preceding symptoms of heart failure.
- In DCM, the heart (especially the left ventricle) is enlarged and the pumping function is diminished. Approximately 40% of cases are familial. Mutations in multiple genes have been described. In some cases it manifests as peripartum cardiomyopathy, and in other cases it may be associated with alcoholism.
- Hypertrophic cardiomyopathy(HCM or HOCM), a genetic disorder caused by various mutations in genes encoding sarcomeric proteins. In HCM the heart muscle is thickened, which can obstruct blood flow and prevent the heart from functioning properly.
- Arrhythmogenic right ventricular cardiomyopathy(ARVC) arises from an electrical disturbance of the heart in which heart muscle is replaced by fibrous scar tissue. The right ventricle is generally most affected, but LV can also be affected.
- Restrictive cardiomyopathy(RCM) is an uncommon cardiomyopathy. The walls of the ventricles are stiff, but may not be thickened, and resist the normal filling of the heart with blood. A rare form of restrictive cardiomyopathy is the obliterative cardiomyopathy in hypereosinophilic syndrome. In this type, the myocardium in the apices of the left and right ventricles becomes thickened and fibrotic, causing a decrease in ventricular volumes.
- Noncompaction cardiomyopathy has been recognized as a separate type since the 1980s. The term refers to a cardiomyopathy where the left ventricle wall has failed to grow properly from birth and has a spongy appearance on echocardiogram. These patients are at risk of heart failure, thromboembolic events and sudden cardiac death
Gross Pathology
-
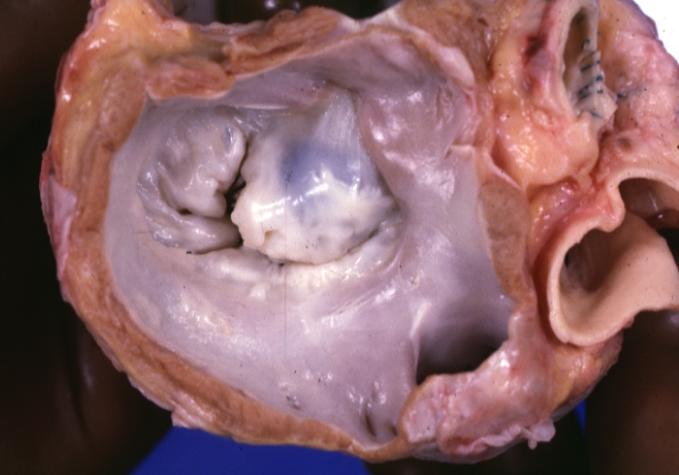
Cardiomyopathy: Gross excellent view of mitral valve from left atrium anterior leaflet appears to balloon a bit into the atrium
-
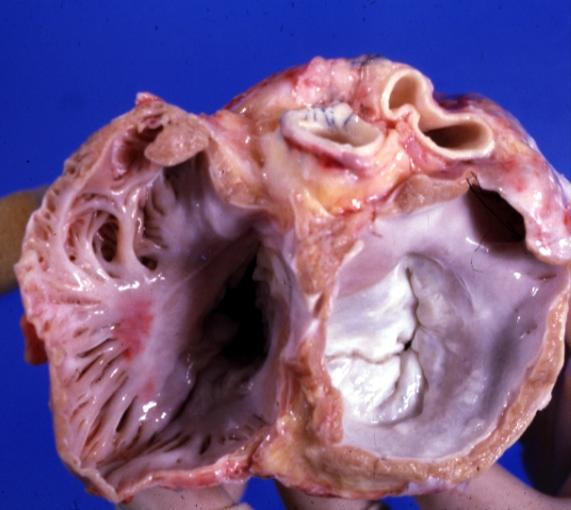
Cardiomyopathy: Gross excellent view of mitral and tricuspid valves from atria, appear normal anatomy.


-
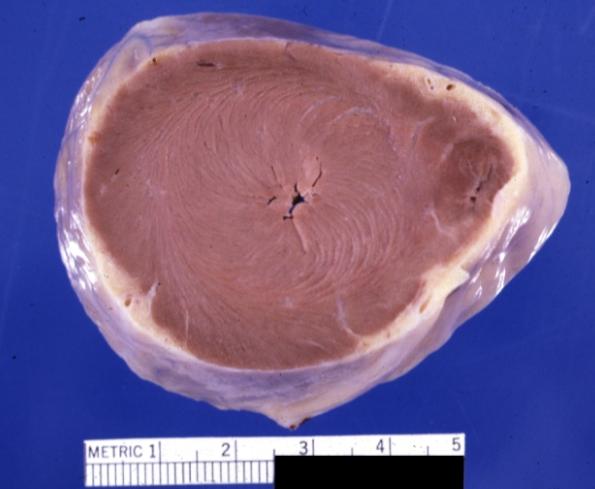
Cardiomyopathy: Gross apical slice of left and right ventricles concentric hypertrophy with cavitary obliteration sudden unexpected death obstructive cardiomyopathy
-
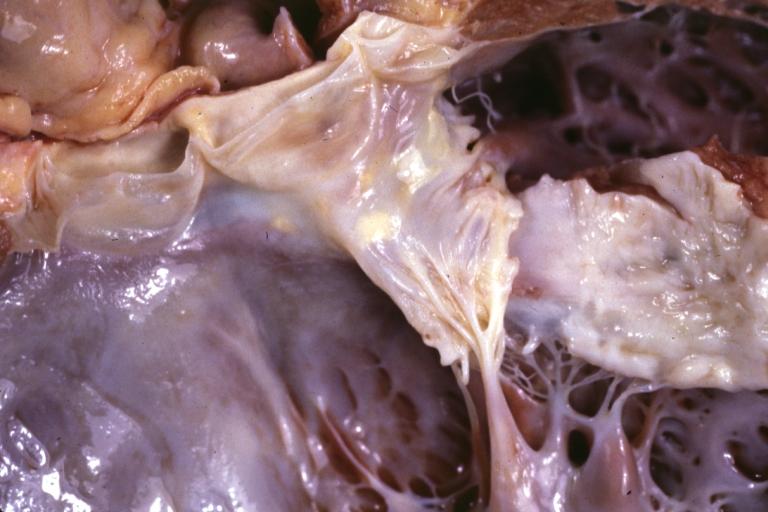
Dilated Cardiomyopathy: Gross natural color close-up view of heart surgically removed for a transplantation shows aortic valve and anterior leaflet of mitral valve with cholesterol deposits endocardium of left ventricle is diffusely thickened


-
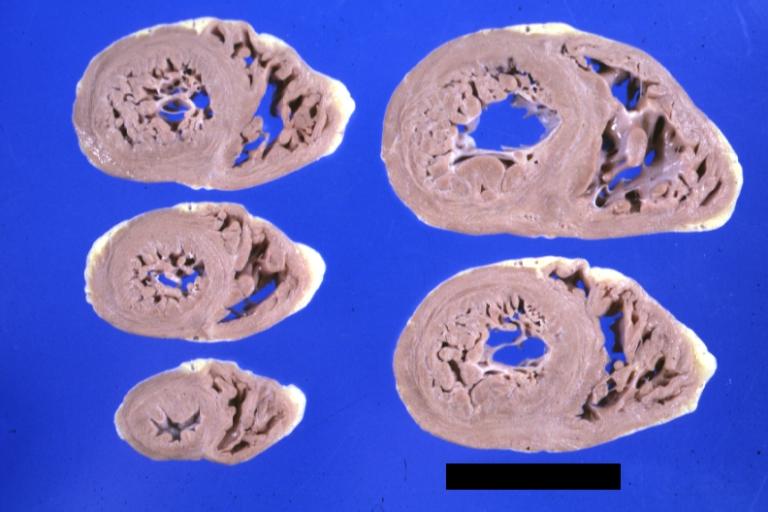
Cardiomyopathy: Gross montage of ventricular slices showing hypertrophy and about normal ventricular lumen size a hypertrophic non-dilated cardiomyopathy
-
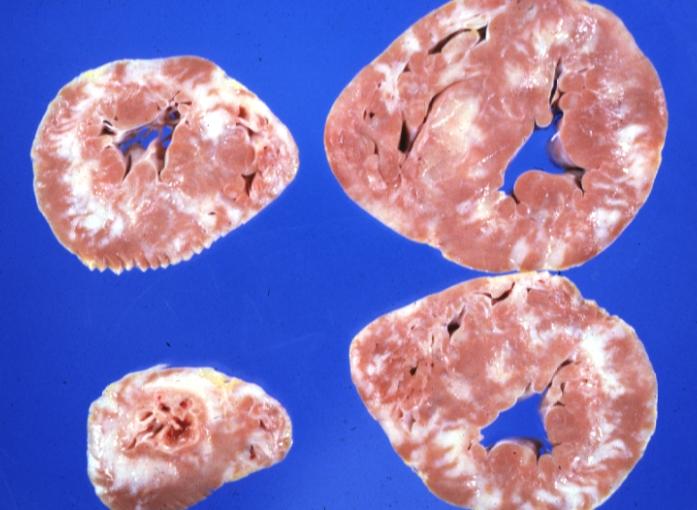
Cardiomyopathy: Gross ventricular slices hypertrophy and extensive myocardial fibrosis a unique case of global fiber disarray with atrophy and fibrosis


-
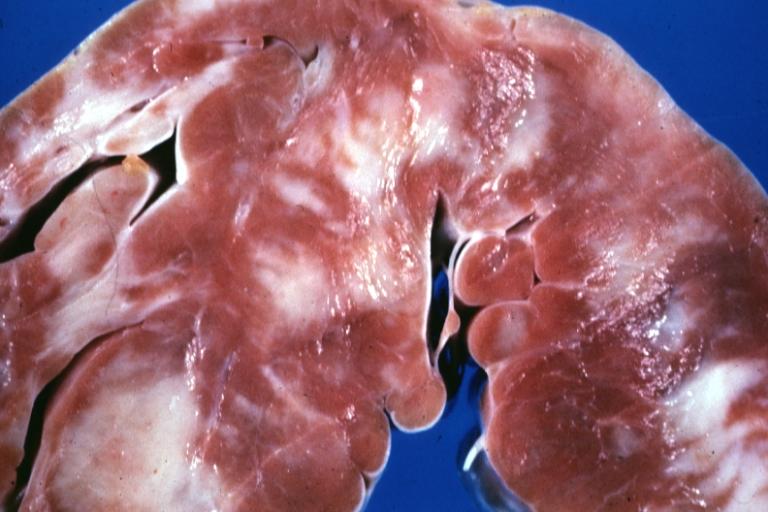
Cardiomyopathy: Gross close up view of a ventricle slice
-
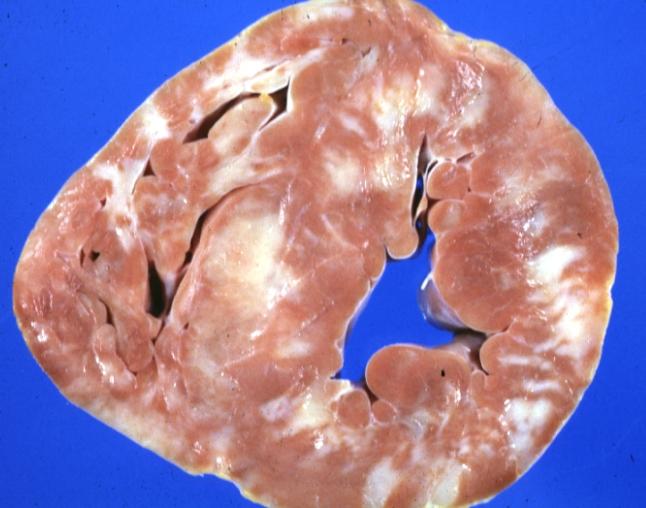
Cardiomyopathy: Gross excellent ventricular slice with hypertrophy and fibrosis a unique case of global fiber disarray with hypertrophy then atrophy and then fibrosis


-
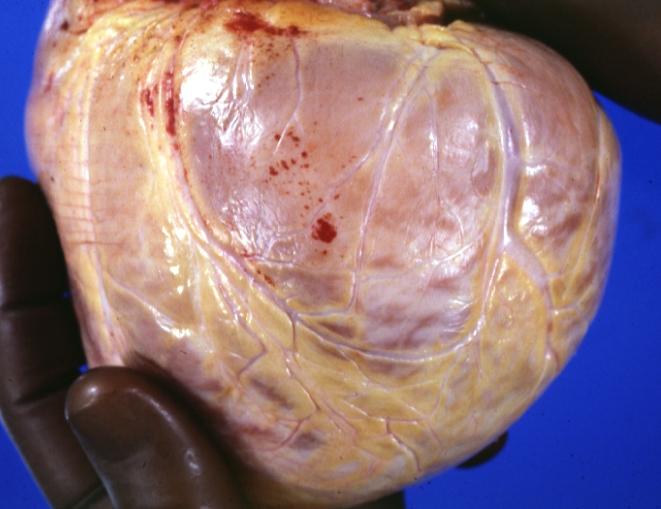
Cardiomyopathy: Gross external view of globular heart with patchy fibrosis seen through epicardium
-
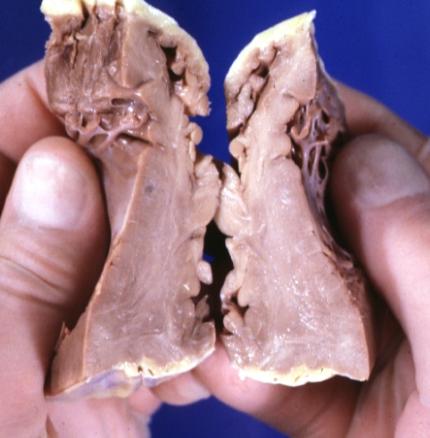
Cardiomyopathy: Gross interventricular septum showing asymmetrical hypertrophy in posterior septum


-
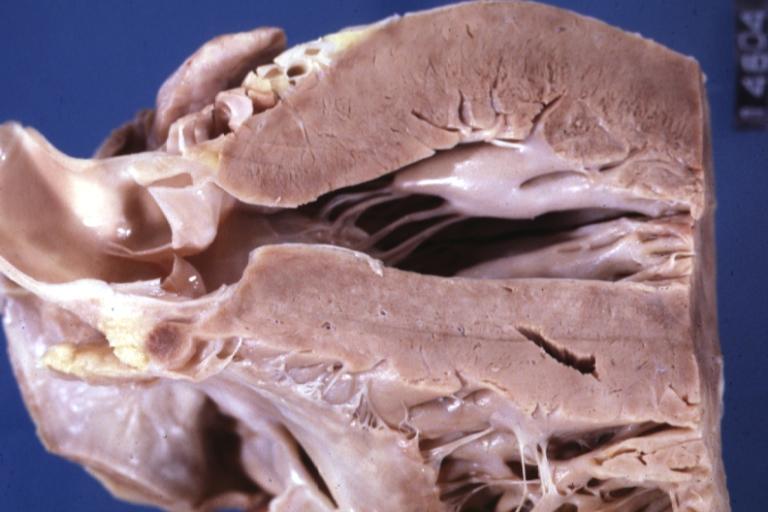
Cardiomyopathy: Gross hypertrophic cardiomyopathy obstructive excellent section through left ventricle outlet to show subvalvular narrowing case of sudden death in a 27 yo male playing basketball no history of disease
-
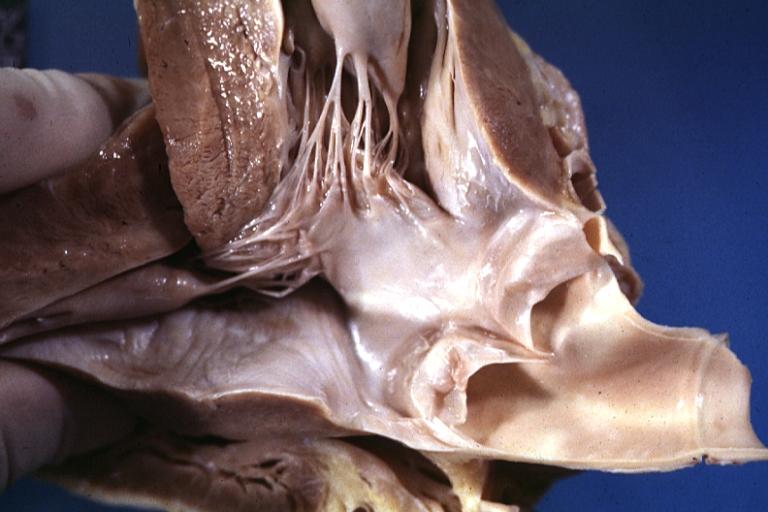
Cardiomyopathy: Gross obstructive cardiomyopathy showing aorta outflow tract with marked endocardial thickening mitral valve appears normal (Same case as previous one)


-
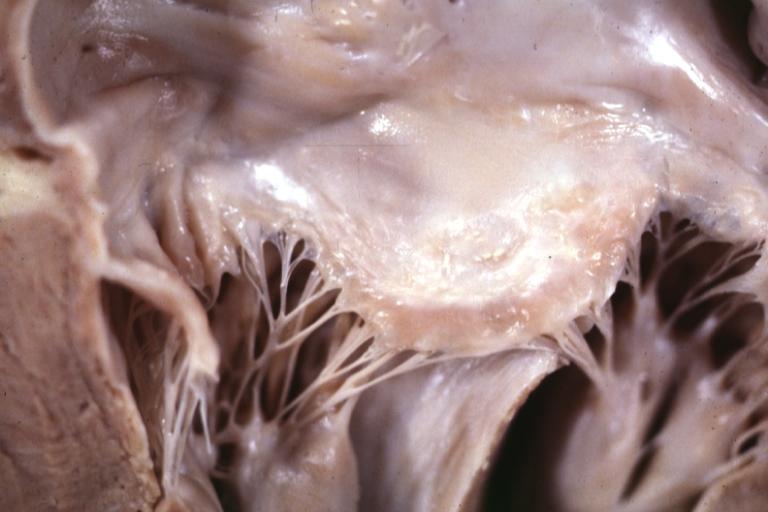
Cardiomyopathy: Gross excellent view of mitral valve atrial surface showing thickening which is fibrous in body of valve and myxoid at area of free margin changes presumed secondary to insufficiency due to anterior motion
-
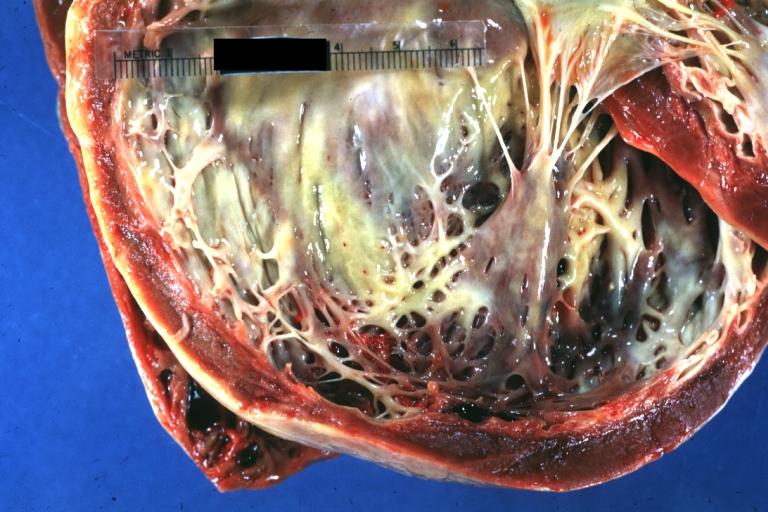
Cardiomyopathy: Gross dilated left ventricle with marked endocardial thickening this is what has been called adult fibroelastosis


-
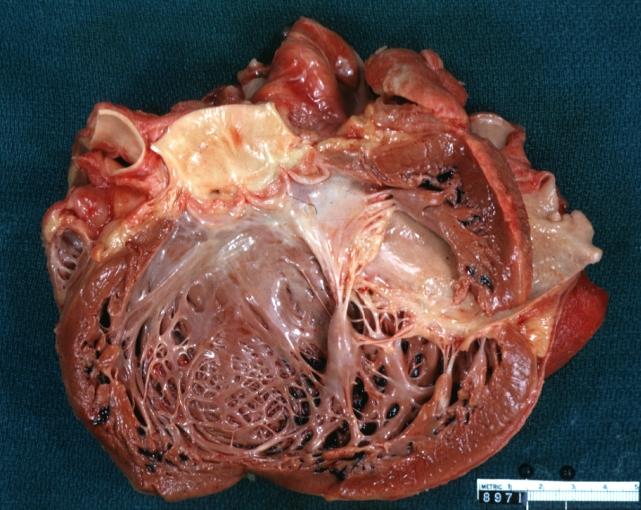
Dilated Cardiomyopathy: Gross good example huge dilated left ventricle
-
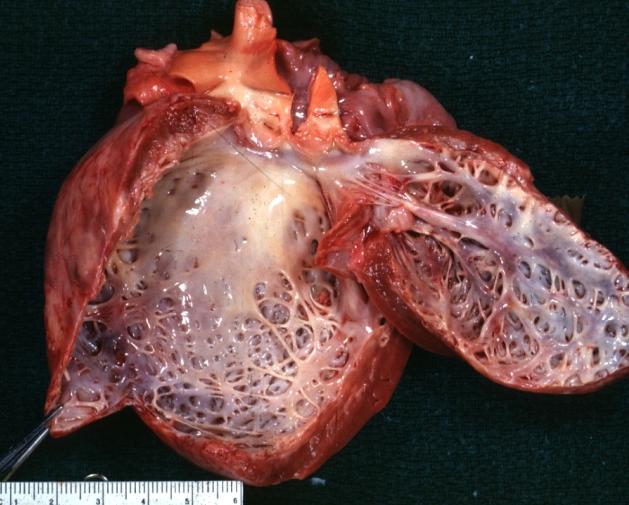
Dilated Cardiomyopathy: Gross dilated left ventricle with marked endocardial sclerosis (an excellent example)


-
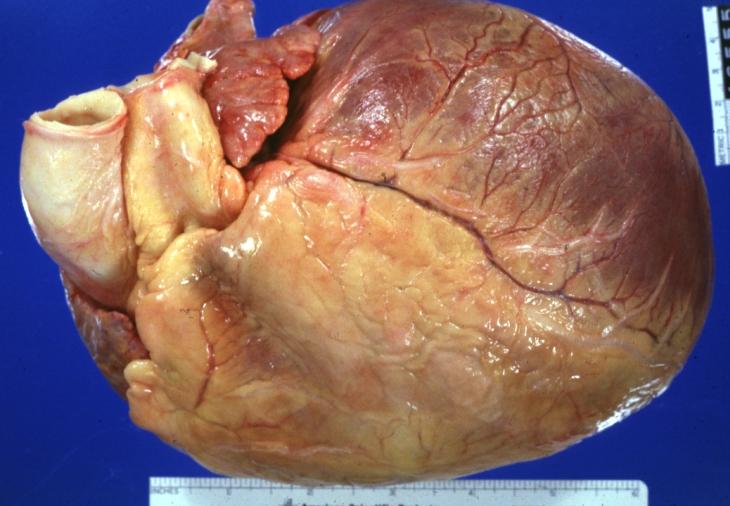
Cardiomyopathy: Gross intact globular shaped heart
-
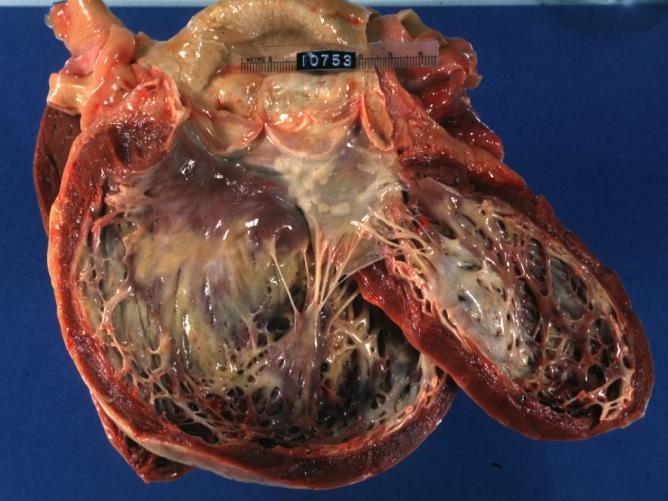
Dilated Cardiomyopathy: Gross opened left ventricle dilated with endocardial thickening good example


-
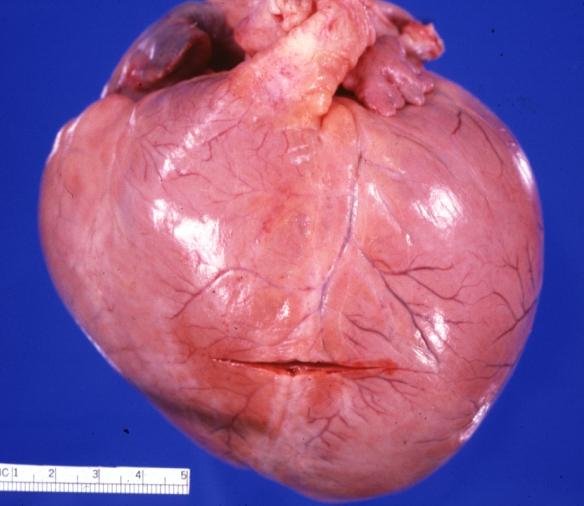
Cardiomyopathy: Gross globular heart external view 10 year old girl with sickle cell anemia
-
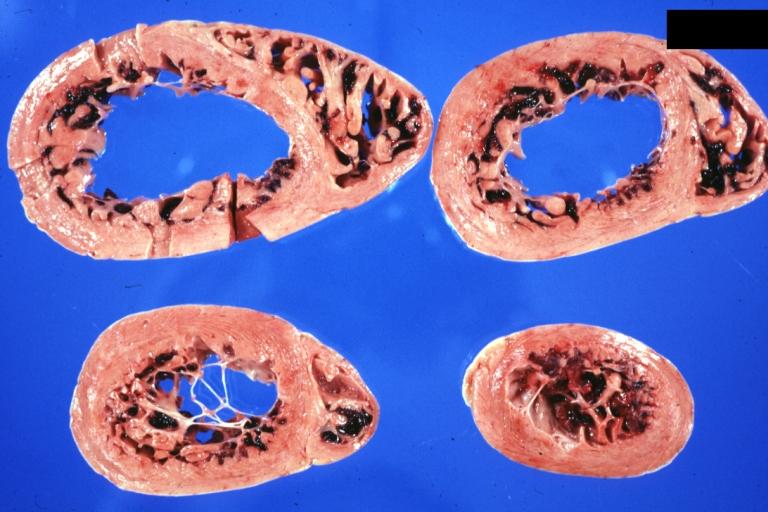
Cardiomyopathy: Gross horizontal sections of ventricles dilation type 10 year old girl with sickle cell anemia


-
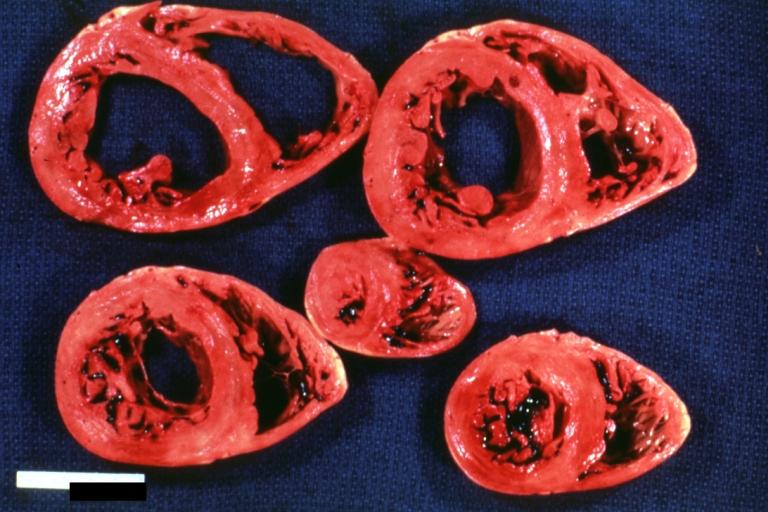
Cardiomyopathy: Intermediate between hypertrophic and dilated
-
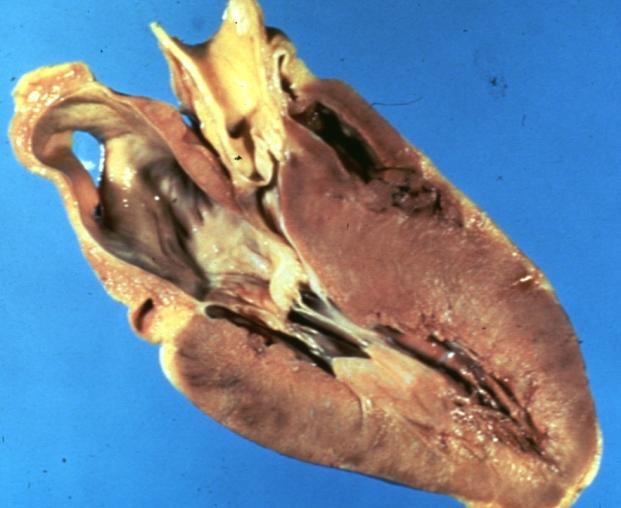
Cardiomyopathy Asymmetrical Septal Hypertrophy


-
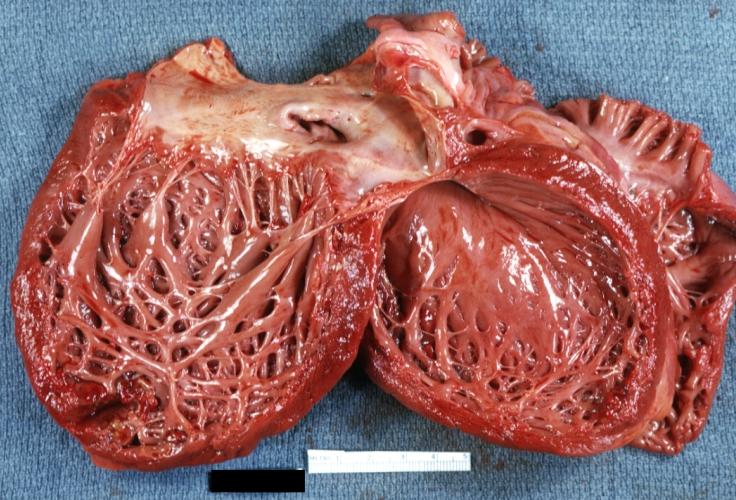
Dilated Cardiomyopathy: Gross opened globular left ventricle natural color (very good example)
-
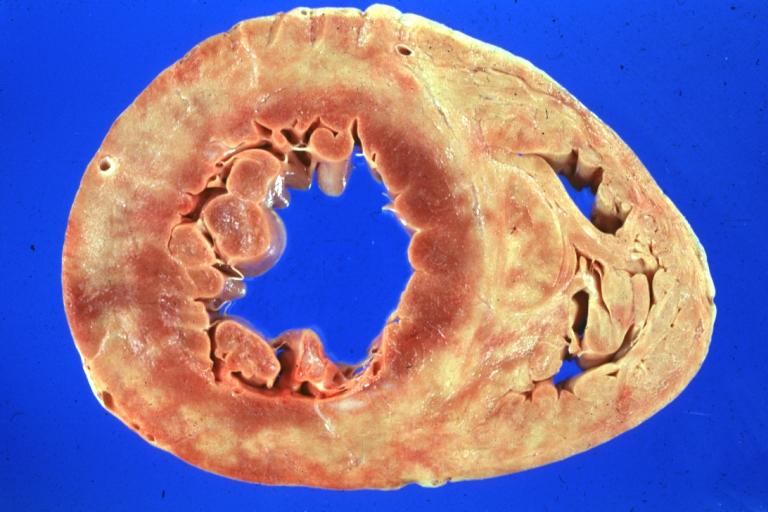
Diabetic Cardiomyopathy: Gross natural color moderately hypertrophied heart shown in horizontal section hyperemic subendocardium has no microscopic lesion long standing type I diabetic patient, no significant coronary artery lesions, congestive heart failure


-
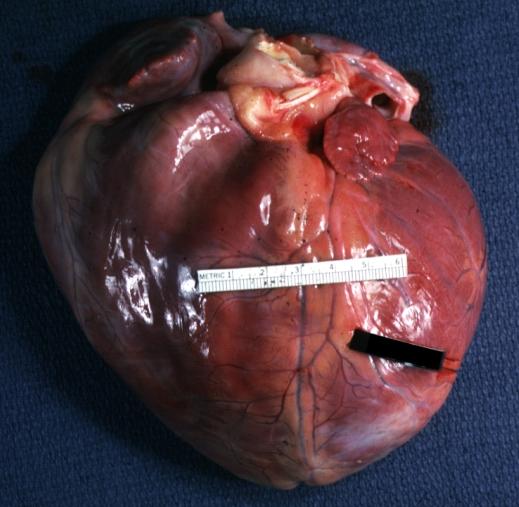
Dilated Cardiomyopathy: Gross natural color external view globular heart 500 gm 24yo female seven pregnancies

Microscopic Pathology
-
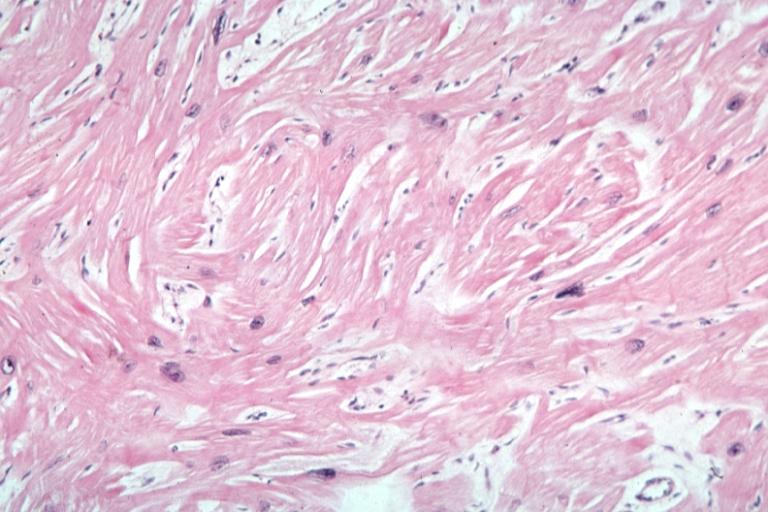
Cardiomyopathy: Micro H&E high mag excellent example myofiber disarray
-
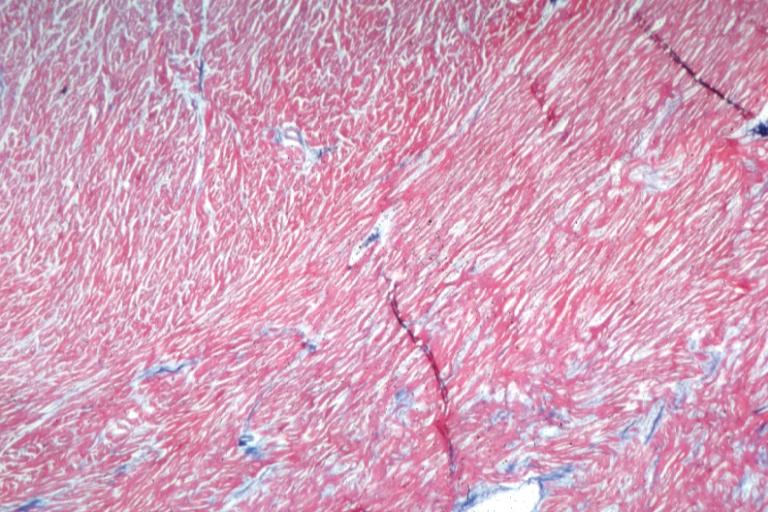
Cardiomyopathy: Micro H&E low mag interventricular septum at junction of normal myofiber orientation with asymmetrical hypertrophy (an excellent example)


-
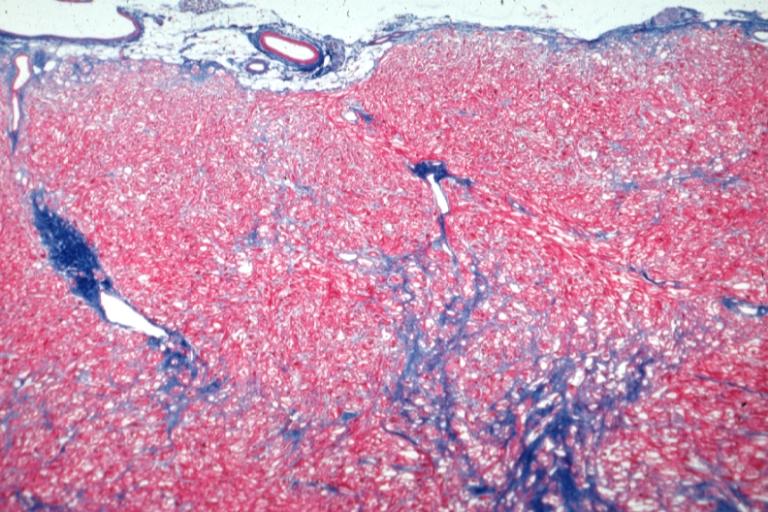
Cardiomyopathy: Micro trichrome low mag bizarre vacuolated fibers with disarray and focal fibrosis excellent low mag epicardial surface
-
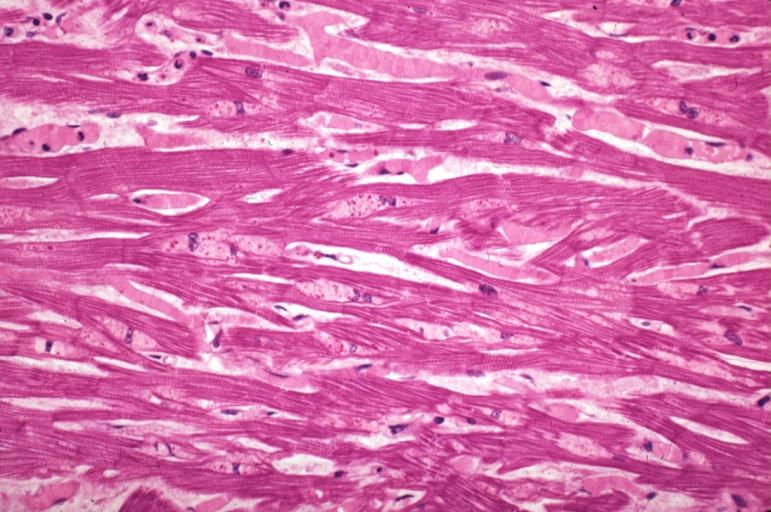
Alcoholic Cardiomyopathy: Micro plastic section lipid in perinuclear area loss of myofibrils

