* Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
* Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
=====MALIGNANCIES=====
=====MALIGNANCIES=====
* Lymphoma and other malignancies have been observed in patients treated with XELJANZ. Epstein Barr Virus- associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with XELJANZ and concomitant immunosuppressive medications
* Lymphoma and other malignancies have been observed in patients treated with XELJANZ. Epstein Barr Virus- associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with XELJANZ and concomitant immunosuppressive medications
|fdaLIADAdult======Rheumatoid Arthritis=====
|fdaLIADAdult======Rheumatoid Arthritis=====
* XELJANZ (tofacitinib) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs).
* XELJANZ (tofacitinib) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs).
Line 33:
Line 33:
* Coadministration of XELJANZ with potent inducers of CYP3A4 (e.g., rifampin) may result in loss of or reduced clinical response to XELJANZ.
* Coadministration of XELJANZ with potent inducers of CYP3A4 (e.g., rifampin) may result in loss of or reduced clinical response to XELJANZ.
: [[File:T 01.png|thumb|none|600px|This image is provided by the National Library of Medicine.]]
: [[File:T 01.png|thumb|none|600px|This image is provided by the National Library of Medicine.]]
|offLabelAdultGuideSupport=There is limited information regarding <i>Off-Label Guideline-Supported Use</i> of {{PAGENAME}} in adult patients.
|offLabelAdultGuideSupport=
There is limited information regarding <i>Off-Label Guideline-Supported Use</i> of {{PAGENAME}} in adult patients.
<!--Non–Guideline-Supported Use (Adult)-->
<!--Non–Guideline-Supported Use (Adult)-->
|offLabelAdultNoGuideSupport=
|offLabelAdultNoGuideSupport=There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in adult patients.
There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in adult patients.
<!--Pediatric Indications and Dosage-->
<!--Pediatric Indications and Dosage-->
<!--FDA-Labeled Indications and Dosage (Pediatric)-->
<!--FDA-Labeled Indications and Dosage (Pediatric)-->
|fdaLIADPed=
|fdaLIADPed=There is limited information regarding <i>FDA-Labeled Use</i> of {{PAGENAME}} in pediatric patients.
There is limited information regarding <i>FDA-Labeled Use</i> of {{PAGENAME}} in pediatric patients.
<!--Off-Label Use and Dosage (Pediatric)-->
<!--Off-Label Use and Dosage (Pediatric)-->
Line 54:
Line 49:
<!--Non–Guideline-Supported Use (Pediatric)-->
<!--Non–Guideline-Supported Use (Pediatric)-->
|offLabelPedNoGuideSupport=
|offLabelPedNoGuideSupport=There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in pediatric patients.
There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in pediatric patients.
<!--Contraindications-->
<!--Contraindications-->
|contraindications=
|contraindications=<!--Warnings-->
<!--Warnings-->
|warnings======Serious Infections=====
|warnings======Serious Infections=====
* Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, or other opportunistic pathogens have been reported in rheumatoid arthritis patients receiving XELJANZ. The most common serious infections reported with XELJANZ included pneumonia, cellulitis, herpes zoster and urinary tract infection.
* Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, or other opportunistic pathogens have been reported in rheumatoid arthritis patients receiving XELJANZ. The most common serious infections reported with XELJANZ included pneumonia, cellulitis, herpes zoster and urinary tract infection.
Line 153:
Line 145:
* Mean HDL cholesterol increased by 10% in the XELJANZ 5 mg twice daily arm and 12% in the XELJANZ 10 mg twice daily arm.
* Mean HDL cholesterol increased by 10% in the XELJANZ 5 mg twice daily arm and 12% in the XELJANZ 10 mg twice daily arm.
* Mean LDL/HDL ratios were essentially unchanged in XELJANZ-treated patients.
* Mean LDL/HDL ratios were essentially unchanged in XELJANZ-treated patients.
In a controlled clinical trial, elevations in LDL cholesterol and ApoB decreased to pretreatment levels in response to statin therapy.
* In a controlled clinical trial, elevations in LDL cholesterol and ApoB decreased to pretreatment levels in response to statin therapy.
* In the long-term safety population, elevations in lipid parameters remained consistent with what was seen in the controlled clinical trials.
* In the long-term safety population, elevations in lipid parameters remained consistent with what was seen in the controlled clinical trials.
======Serum Creatinine======
======Serum Creatinine======
Line 180:
Line 172:
* General disorders and administration site conditions
* General disorders and administration site conditions
* Tofacitinib exposure is increased when XELJANZ is coadministered with potent inhibitors of cytochrome P450 (CYP) 3A4 (e.g., ketoconazole).
* Tofacitinib exposure is increased when XELJANZ is coadministered with potent inhibitors of cytochrome P450 (CYP) 3A4 (e.g., ketoconazole).
Line 208:
Line 199:
|useInRace=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific racial populations.
|useInRace=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific racial populations.
|useInRenalImpair=* No dose adjustment is required in patients with mild renal impairment. XELJANZ dose should be reduced to 5 mg once daily in patients with moderate and severe renal impairment. In clinical trials, XELJANZ was not evaluated in rheumatoid arthritis patients with baseline creatinine clearance values (estimated by the Cockroft-Gault equation) less than 40 mL/min.
|useInRenalImpair=* No dose adjustment is required in patients with mild renal impairment. XELJANZ dose should be reduced to 5 mg once daily in patients with moderate and severe renal impairment. In clinical trials, XELJANZ was not evaluated in rheumatoid arthritis patients with baseline creatinine clearance values (estimated by the Cockroft-Gault equation) less than 40 mL/min.
|useInHepaticImpair=* Treatment with XELJANZ is not recommended in patients with severe hepatic impairment. No dose adjustment is required in patients with mild hepatic impairment. XELJANZ dose should be reduced to 5 mg once daily in patients with moderate hepatic impairment. The safety and efficacy of XELJANZ have not been studied in patients with severe hepatic impairment or in patients with positive hepatitis B virus or hepatitis C virus serology
|useInHepaticImpair=* Treatment with XELJANZ is not recommended in patients with severe hepatic impairment. No dose adjustment is required in patients with mild hepatic impairment. XELJANZ dose should be reduced to 5 mg once daily in patients with moderate hepatic impairment. The safety and efficacy of XELJANZ have not been studied in patients with severe hepatic impairment or in patients with positive hepatitis B virus or hepatitis C virus serology
|useInReproPotential=There is no FDA guidance on the use of {{PAGENAME}} in women of reproductive potentials and males.
|useInReproPotential=There is no FDA guidance on the use of {{PAGENAME}} in women of reproductive potentials and males.
|useInImmunocomp=There is no FDA guidance one the use of {{PAGENAME}} in patients who are immunocompromised.
|useInImmunocomp=There is no FDA guidance one the use of {{PAGENAME}} in patients who are immunocompromised.
Line 215:
Line 206:
|othersTitle=Nonteratogenic effects:
|othersTitle=Nonteratogenic effects:
|administration=* Oral
|administration=* Oral
|monitoring=* Laboratory monitoring –Recommended due to potential changes in lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids.
|monitoring=* Laboratory monitoring –Recommended due to potential changes in lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids.
* Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ. XELJANZ should be interrupted if a patient develops a serious infection, an opportunistic infection, or sepsis.
* Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ. XELJANZ should be interrupted if a patient develops a serious infection, an opportunistic infection, or sepsis.
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
WARNING: SERIOUS INFECTIONS AND MALIGNANCY
See full prescribing information for complete Boxed Warning.
* Patients treated with XELJANZ are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt XELJANZ until the infection is controlled.
Reported infections include:
Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before XELJANZ use and during therapy. Treatment for latent infection should be initiated prior to XELJANZ use.
Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
Bacterial, viral, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with XELJANZ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with XELJANZ. Epstein Barr Virus- associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with XELJANZ and concomitant immunosuppressive medications
Overview
Tofacitinib is a anti rheumatic that is FDA approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis. There is a Black Box Warning for this drug as shown here. Common adverse reactions include anemia, hepatic steatosis, pruritis, tendonitis, pyrexia.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Rheumatoid Arthritis
XELJANZ (tofacitinib) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs).
XELJANZ should not be used in combination with biologic DMARDs or with potent immunosuppressants such as azathioprine and cyclosporine.
The recommended dose of XELJANZ is 5 mg twice daily.
XELJANZ dosage should be reduced to 5 mg once daily in patients:
with moderate or severe renal insufficiency
with moderate hepatic impairment
receiving potent inhibitors of Cytochrome P450 3A4 (CYP3A4) (e.g., ketoconazole)
receiving one or more concomitant medications that result in both moderate inhibition of CYP3A4 and potent inhibition of CYP2C19 (e.g., fluconazole).
General Considerations for Administration
XELJANZ should not be used in patients with severe hepatic impairment.
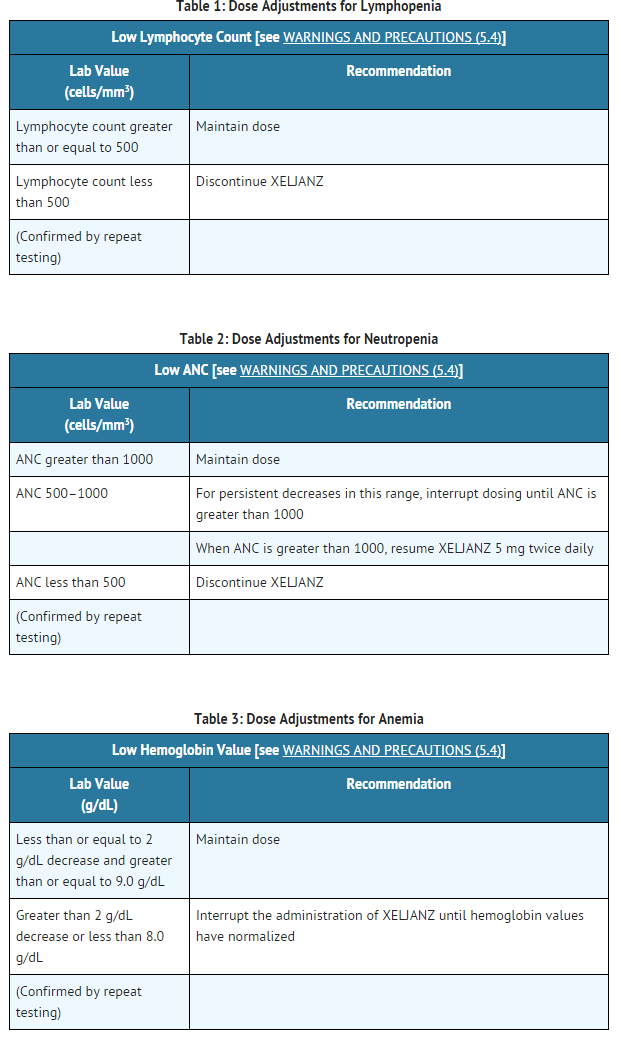
It is recommended that XELJANZ not be initiated in patients with a lymphocyte count less than 500 cells/mm3, an absolute neutrophil count (ANC) less than 1000 cells/mm3, or who have hemoglobin levels less than 9 g/dL.
Coadministration of XELJANZ with potent inducers of CYP3A4 (e.g., rifampin) may result in loss of or reduced clinical response to XELJANZ.
This image is provided by the National Library of Medicine.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Tofacitinib in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Tofacitinib in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Tofacitinib in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Tofacitinib in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Tofacitinib in pediatric patients.
Contraindications
There is limited information regarding Tofacitinib Contraindications in the drug label.
Warnings
WARNING: SERIOUS INFECTIONS AND MALIGNANCY
See full prescribing information for complete Boxed Warning.
* Patients treated with XELJANZ are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt XELJANZ until the infection is controlled.
Reported infections include:
Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before XELJANZ use and during therapy. Treatment for latent infection should be initiated prior to XELJANZ use.
Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
Bacterial, viral, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with XELJANZ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with XELJANZ. Epstein Barr Virus- associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with XELJANZ and concomitant immunosuppressive medications
Serious Infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, or other opportunistic pathogens have been reported in rheumatoid arthritis patients receiving XELJANZ. The most common serious infections reported with XELJANZ included pneumonia, cellulitis, herpes zoster and urinary tract infection.
Among opportunistic infections, tuberculosis and other mycobacterial infections, cryptococcus, esophageal candidiasis, pneumocystosis, multidermatomal herpes zoster, cytomegalovirus, and BK virus were reported with XELJANZ. Some patients have presented with disseminated rather than localized disease, and were often taking concomitant immunomodulating agents such as methotrexate or corticosteroids.
Other serious infections that were not reported in clinical studies may also occur (e.g., histoplasmosis, coccidioidomycosis, and listeriosis).
XELJANZ should not be initiated in patients with an active infection, including localized infections. The risks and benefits of treatment should be considered prior to initiating XELJANZ in patients:
with chronic or recurrent infection
who have been exposed to tuberculosis
with a history of a serious or an opportunistic infection
who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
with underlying conditions that may predispose them to infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ. XELJANZ should be interrupted if a patient develops a serious infection, an opportunistic infection, or sepsis.
A patient who develops a new infection during treatment with XELJANZ should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, and the patient should be closely monitored.
Tuberculosis
Patients should be evaluated and tested for latent or active infection prior to administration of XELJANZ.
Anti-tuberculosis therapy should also be considered prior to administration of XELJANZ in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but who have risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision about whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Patients should be closely monitored for the development of signs and symptoms of tuberculosis, including patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Patients with latent tuberculosis should be treated with standard antimycobacterial therapy before administering XELJANZ.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster), were observed in clinical studies with XELJANZ. The impact of XELJANZ on chronic viral hepatitis reactivation is unknown. Patients who screened positive for hepatitis B or C were excluded from clinical trials.
Malignancy and Lymphoproliferative Disorder
Consider the risks and benefits of XELJANZ treatment prior to initiating therapy in patients with a known malignancy other than a successfully treated non-melanoma skin cancer (NMSC) or when considering continuing XELJANZ in patients who develop a malignancy. Malignancies were observed in clinical studies of XELJANZ.
In the seven controlled rheumatoid arthritis clinical studies, 11 solid cancers and one lymphoma were diagnosed in 3328 patients receiving XELJANZ with or without DMARD, compared to 0 solid cancers and 0 lymphomas in 809 patients in the placebo with or without DMARD group during the first 12 months of exposure. Lymphomas and solid cancers have also been observed in the long-term extension studies in rheumatoid arthritis patients treated with XELJANZ.
In Phase 2B, controlled dose-ranging trials in de-novo renal transplant patients, all of whom received induction therapy with basiliximab, high dose corticosteroids, and mycophenolic acid products, Epstein Barr Virus-associated post-transplant lymphoproliferative disorder was observed in 5 out of 218 patients treated with XELJANZ (2.3%) compared to 0 out of 111 patients treated with cyclosporine.
Gastrointestinal Perforations
Events of gastrointestinal perforation have been reported in clinical studies with XELJANZ in rheumatoid arthritis patients, although the role of JAK inhibition in these events is not known.
XELJANZ should be used with caution in patients who may be at increased risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis). Patients presenting with new onset abdominal symptoms should be evaluated promptly for early identification of gastrointestinal perforation.
Laboratory Parameters
Lymphocytes
Treatment with XELJANZ was associated with initial lymphocytosis at one month of exposure followed by a gradual decrease in mean lymphocyte counts below the baseline of approximately 10% during 12 months of therapy. Lymphocyte counts less than 500 cells/mm3 were associated with an increased incidence of treated and serious infections.
Avoid initiation of XELJANZ treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3). In patients who develop a confirmed absolute lymphocyte count less than 500 cells/mm3 treatment with XELJANZ is not recommended.
Monitor lymphocyte counts at baseline and every 3 months thereafter. For recommended modifications based on lymphocyte counts
Neutrophils
Treatment with XELJANZ was associated with an increased incidence of neutropenia (less than 2000 cells/mm3) compared to placebo.
Avoid initiation of XELJANZ treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3). For patients who develop a persistent ANC of 500–1000 cells/mm3, interrupt XELJANZ dosing until ANC is greater than or equal to 1000 cells/mm3. In patients who develop an ANC less than 500 cells/mm3, treatment with XELJANZ is not recommended.
Monitor neutrophil counts at baseline and after 4–8 weeks of treatment and every 3 months thereafter. For recommended modifications based on ANC results
Hemoglobin
Avoid initiation of XELJANZ treatment in patients with a low hemoglobin level (i.e. less than 9 g/dL). Treatment with XELJANZ should be interrupted in patients who develop hemoglobin levels less than 8 g/dL or whose hemoglobin level drops greater than 2 g/dL on treatment.
Monitor hemoglobin at baseline and after 4–8 weeks of treatment and every 3 months thereafter. For recommended modifications based on hemoglobin results.
Liver Enzymes
Treatment with XELJANZ was associated with an increased incidence of liver enzyme elevation compared to placebo. Most of these abnormalities occurred in studies with background DMARD (primarily methotrexate) therapy.
Routine monitoring of liver tests and prompt investigation of the causes of liver enzyme elevations is recommended to identify potential cases of drug-induced liver injury. If drug-induced liver injury is suspected, the administration of XELJANZ should be interrupted until this diagnosis has been excluded.
Lipids
Treatment with XELJANZ was associated with increases in lipid parameters including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. Maximum effects were generally observed within 6 weeks. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assessment of lipid parameters should be performed approximately 4–8 weeks following initiation of XELJANZ therapy.
Manage patients according to clinical guidelines [e.g., National Cholesterol Educational Program (NCEP)] for the management of hyperlipidemia.
Vaccinations
No data are available on the response to vaccination or on the secondary transmission of infection by live vaccines to patients receiving XELJANZ. Live vaccines should not be given concurrently with XELJANZ.
Update immunizations in agreement with current immunization guidelines prior to initiating XELJANZ therapy.
Adverse Reactions
Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
The following data includes two Phase 2 and five Phase 3 double-blind, controlled, multicenter trials. In these trials, patients were randomized to doses of XELJANZ 5 mg twice daily (292 patients) and 10 mg twice daily (306 patients) monotherapy, XELJANZ 5 mg twice daily (1044 patients) and 10 mg twice daily (1043 patients) in combination with DMARDs (including methotrexate) and placebo (809 patients). All seven protocols included provisions for patients taking placebo to receive treatment with XELJANZ at Month 3 or Month 6 either by patient response (based on uncontrolled disease activity) or by design, so that adverse events cannot always be unambiguously attributed to a given treatment. Therefore some analyses that follow include patients who changed treatment by design or by patient response from placebo to XELJANZ in both the placebo and XELJANZ group of a given interval. Comparisons between placebo and XELJANZ were based on the first 3 months of exposure, and comparisons between XELJANZ 5 mg twice daily and XELJANZ 10 mg twice daily were based on the first 12 months of exposure.
The long-term safety population includes all patients who participated in a double-blind, controlled trial (including earlier development phase studies) and then participated in one of two long-term safety studies. The design of the long-term safety studies allowed for modification of XELJANZ doses according to clinical judgment. This limits the interpretation of the long-term safety data with respect to dose.
Clinical Trial Experience
The most common serious adverse reactions were serious infections.
The proportion of patients who discontinued treatment due to any adverse reaction during the 0 to 3 months exposure in the double-blind, placebo-controlled trials was 4% for patients taking XELJANZ and 3% for placebo-treated patients.
Overall Infections
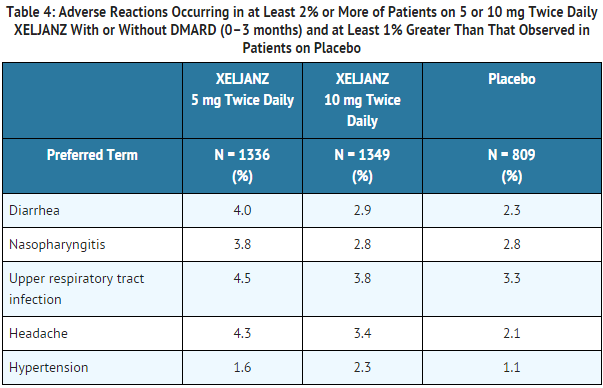
In the seven controlled trials, during the 0 to 3 months exposure, the overall frequency of infections was 20% and 22% in the 5 mg twice daily and 10 mg twice daily groups, respectively, and 18% in the placebo group.
The most commonly reported infections with XELJANZ were upper respiratory tract infections, nasopharyngitis, and urinary tract infections (4%, 3%, and 2% of patients, respectively).
Serious Infections
In the seven controlled trials, during the 0 to 3 months exposure, serious infections were reported in 1 patient (0.5 events per 100 patient-years) who received placebo and 11 patients (1.7 events per 100 patient-years) who received XELJANZ 5 mg or 10 mg twice daily. The rate difference between treatment groups (and the corresponding 95% confidence interval) was 1.1 (-0.4, 2.5) events per 100 patient-years for the combined 5 mg twice daily and 10 mg twice daily XELJANZ group minus placebo.
In the seven controlled trials, during the 0 to 12 months exposure, serious infections were reported in 34 patients (2.7 events per 100 patient-years) who received 5 mg twice daily of XELJANZ and 33 patients (2.7 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was -0.1 (-1.3, 1.2) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ.
The most common serious infections included pneumonia, cellulitis, herpes zoster, and urinary tract infection.
Tuberculosis
In the seven controlled trials, during the 0 to 3 months exposure, tuberculosis was not reported in patients who received placebo, 5 mg twice daily of XELJANZ, or 10 mg twice daily of XELJANZ.
In the seven controlled trials, during the 0 to 12 months exposure, tuberculosis was reported in 0 patients who received 5 mg twice daily of XELJANZ and 6 patients (0.5 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was 0.5 (0.1, 0.9) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ.
Cases of disseminated tuberculosis were also reported. The median XELJANZ exposure prior to diagnosis of tuberculosis was 10 months (range from 152 to 960 days)
Opportunistic Infections (excluding tuberculosis)
In the seven controlled trials, during the 0 to 3 months exposure, opportunistic infections were not reported in patients who received placebo, 5 mg twice daily of XELJANZ, or 10 mg twice daily of XELJANZ.
In the seven controlled trials, during the 0 to 12 months exposure, opportunistic infections were reported in 4 patients (0.3 events per 100 patient-years) who received 5 mg twice daily of XELJANZ and 4 patients (0.3 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was 0 (-0.5, 0.5) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ.
The median XELJANZ exposure prior to diagnosis of an opportunistic infection was 8 months (range from 41 to 698 days).
Malignancy
In the seven controlled trials, during the 0 to 3 months exposure, malignancies excluding NMSC were reported in 0 patients who received placebo and 2 patients (0.3 events per 100 patient-years) who received either XELJANZ 5 mg or 10 mg twice daily. The rate difference between treatment groups (and the corresponding 95% confidence interval) was 0.3 (-0.1, 0.7) events per 100 patient-years for the combined 5 mg and 10 mg twice daily XELJANZ group minus placebo.
In the seven controlled trials, during the 0 to 12 months exposure, malignancies excluding NMSC were reported in 5 patients (0.4 events per 100 patient-years) who received 5 mg twice daily of XELJANZ and 7 patients (0.6 events per 100 patient-years) who received 10 mg twice daily of XELJANZ. The rate difference between XELJANZ doses (and the corresponding 95% confidence interval) was 0.2 (-0.4, 0.7) events per 100 patient-years for 10 mg twice daily XELJANZ minus 5 mg twice daily XELJANZ. One of these malignancies was a case of lymphoma that occurred during the 0 to 12 month period in a patient treated with XELJANZ 10 mg twice daily.
The most common types of malignancy, including malignancies observed during the long-term extension, were lung and breast cancer, followed by gastric, colorectal, renal cell, prostate cancer, lymphoma, and malignant melanoma.
Laboratory Tests
Lymphocytes
In the controlled clinical trials, confirmed decreases in lymphocyte counts below 500 cells/mm3 occurred in 0.04% of patients for the 5 mg twice daily and 10 mg twice daily XELJANZ groups combined during the first 3 months of exposure.
Confirmed lymphocyte counts less than 500 cells/mm3 were associated with an increased incidence of treated and serious infections.
Neutrophils
In the controlled clinical trials, confirmed decreases in ANC below 1000 cells/mm3 occurred in 0.07% of patients for the 5 mg twice daily and 10 mg twice daily XELJANZ groups combined during the first 3 months of exposure.
There were no confirmed decreases in ANC below 500 cells/mm3 observed in any treatment group.
There was no clear relationship between neutropenia and the occurrence of serious infections.
In the long-term safety population, the pattern and incidence of confirmed decreases in ANC remained consistent with what was seen in the controlled clinical trials.
Liver Enzyme Tests
Confirmed increases in liver enzymes greater than 3 times the upper limit of normal (3× ULN) were observed in patients treated with XELJANZ. In patients experiencing liver enzyme elevation, modification of treatment regimen, such as reduction in the dose of concomitant DMARD, interruption of XELJANZ, or reduction in XELJANZ dose, resulted in decrease or normalization of liver enzymes.
In the controlled monotherapy trials (0–3 months), no differences in the incidence of ALT or AST elevations were observed between the placebo, and XELJANZ 5 mg, and 10 mg twice daily groups.
In the controlled background DMARD trials (0–3 months), ALT elevations greater than 3× ULN were observed in 1.0%, 1.3% and 1.2% of patients receiving placebo, 5 mg, and 10 mg twice daily, respectively. In these trials, AST elevations greater than 3× ULN were observed in 0.6%, 0.5% and 0.4% of patients receiving placebo, 5 mg, and 10 mg twice daily, respectively.
One case of drug-induced liver injury was reported in a patient treated with XELJANZ 10 mg twice daily for approximately 2.5 months. The patient developed symptomatic elevations of AST and ALT greater than 3× ULN and bilirubin elevations greater than 2× ULN, which required hospitalizations and a liver biopsy.
Lipids
In the controlled clinical trials, dose-related elevations in lipid parameters (total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides) were observed at one month of exposure and remained stable thereafter. Changes in lipid parameters during the first 3 months of exposure in the controlled clinical trials are summarized below:
Mean LDL cholesterol increased by 15% in the XELJANZ 5 mg twice daily arm and 19% in the XELJANZ 10 mg twice daily arm.
Mean HDL cholesterol increased by 10% in the XELJANZ 5 mg twice daily arm and 12% in the XELJANZ 10 mg twice daily arm.
Mean LDL/HDL ratios were essentially unchanged in XELJANZ-treated patients.
In a controlled clinical trial, elevations in LDL cholesterol and ApoB decreased to pretreatment levels in response to statin therapy.
In the long-term safety population, elevations in lipid parameters remained consistent with what was seen in the controlled clinical trials.
Serum Creatinine
In the controlled clinical trials, dose-related elevations in serum creatinine were observed with XELJANZ treatment. The mean increase in serum creatinine was <0.1 mg/dL in the 12-month pooled safety analysis; however with increasing duration of exposure in the long-term extensions, up to 2% of patients were discontinued from XELJANZ treatment due to the protocol-specified discontinuation criterion of an increase in creatinine by more than 50% of baseline. The clinical significance of the observed serum creatinine elevations is unknown.
Other Adverse Reactions
Adverse reactions occurring in 2% or more of patients on 5 mg twice daily or 10 mg twice daily XELJANZ and at least 1% greater than that observed in patients on placebo with or without DMARD are summarized in Table 4.
This image is provided by the National Library of Medicine.
General disorders and administration site conditions
Pyrexia, fatigue, peripheral edema
Drug Interactions
Potent CYP3A4 Inhibitors
Tofacitinib exposure is increased when XELJANZ is coadministered with potent inhibitors of cytochrome P450 (CYP) 3A4 (e.g., ketoconazole).
Moderate CYP3A4 and Potent CYP2C19 Inhibitors
Tofacitinib exposure is increased when XELJANZ is coadministered with medications that result in both moderate inhibition of CYP3A4 and potent inhibition of CYP2C19 (e.g., fluconazole).
Potent CYP3A4 Inducers
Tofacitinib exposure is decreased when XELJANZ is coadministered with potent CYP3A4 inducers (e.g., rifampin) .
Immunosuppressive Drugs
There is a risk of added immunosuppression when XELJANZ is coadministered with potent immunosuppressive drugs (e.g., azathioprine, tacrolimus, cyclosporine). Combined use of multiple-dose XELJANZ with potent immunosuppressives has not been studied in rheumatoid arthritis.
There are no adequate and well-controlled studies in pregnant women. XELJANZ should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Tofacitinib has been shown to be fetocidal and teratogenic in rats and rabbits when given at exposures 146 times and 13 times, respectively, the maximum recommended human dose (MRHD).
In a rat embryofetal developmental study, tofacitinib was teratogenic at exposure levels approximately 146 times the MRHD (on an AUC basis at oral doses of 100 mg/kg/day). Teratogenic effects consisted of external and soft tissue malformations of anasarca and membranous ventricular septal defects, respectively, and skeletal malformations or variations (absent cervical arch; bent femur, fibula, humerus, radius, scapula, tibia, and ulna; sternoschisis; absent rib; misshapen femur; branched rib; fused rib; fused sternebra; and hemicentric thoracic centrum).
In addition, there was an increase in post-implantation loss, consisting of early and late resorptions, resulting in a reduced number of viable fetuses. Mean fetal body weight was reduced. No developmental toxicity was observed in rats at exposure levels approximately 58 times the MRHD (on an AUC basis at oral doses of 30 mg/kg/day). In the rabbit embryofetal developmental study, tofacitinib was teratogenic at exposure levels approximately 13 times the MRHD (on an AUC basis at oral doses of 30 mg/kg/day) in the absence of signs of maternal toxicity.
Teratogenic effects included thoracogastroschisis, omphalocele, membranous ventricular septal defects, and cranial/skeletal malformations (microstomia, microphthalmia), mid-line and tail defects. In addition, there was an increase in post-implantation loss associated with late resorptions. No developmental toxicity was observed in rabbits at exposure levels approximately 3 times the MRHD (on an AUC basis at oral doses of 10 mg/kg/day).
Nonteratogenic effects
In a peri- and postnatal rat study, there were reductions in live litter size, postnatal survival, and pup body weights at exposure levels approximately 73 times the MRHD (on an AUC basis at oral doses of 50 mg/kg/day). There was no effect on behavioral and learning assessments, sexual maturation or the ability of the F1 generation rats to mate and produce viable F2 generation fetuses in rats at exposure levels approximately 17 times the MRHD (on an AUC basis at oral doses of 10 mg/kg/day).
Pregnancy registry has been established to monitor the outcomes of pregnant women exposed to XELJAN
Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Tofacitinib in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Tofacitinib during labor and delivery.
Nursing Mothers
Tofacitinib was secreted in milk of lactating rats. It is not known whether tofacitinib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from tofacitinib, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug for the mother.
Pediatric Use
The safety and effectiveness of XELJANZ in pediatric patients have not been established.
Geriatic Use
Of the 3315 patients who enrolled in Studies I to V, a total of 505 rheumatoid arthritis patients were 65 years of age and older, including 71 patients 75 years and older. The frequency of serious infection among XELJANZ-treated subjects 65 years of age and older was higher than among those under the age of 65. As there is a higher incidence of infections in the elderly population in general, caution should be used when treating the elderly.
Gender
There is no FDA guidance on the use of Tofacitinib with respect to specific gender populations.
Race
There is no FDA guidance on the use of Tofacitinib with respect to specific racial populations.
Renal Impairment
No dose adjustment is required in patients with mild renal impairment. XELJANZ dose should be reduced to 5 mg once daily in patients with moderate and severe renal impairment. In clinical trials, XELJANZ was not evaluated in rheumatoid arthritis patients with baseline creatinine clearance values (estimated by the Cockroft-Gault equation) less than 40 mL/min.
Hepatic Impairment
Treatment with XELJANZ is not recommended in patients with severe hepatic impairment. No dose adjustment is required in patients with mild hepatic impairment. XELJANZ dose should be reduced to 5 mg once daily in patients with moderate hepatic impairment. The safety and efficacy of XELJANZ have not been studied in patients with severe hepatic impairment or in patients with positive hepatitis B virus or hepatitis C virus serology
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Tofacitinib in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Tofacitinib in patients who are immunocompromised.
Nonteratogenic effects:
Administration and Monitoring
Administration
Oral
Monitoring
Laboratory monitoring –Recommended due to potential changes in lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with XELJANZ. XELJANZ should be interrupted if a patient develops a serious infection, an opportunistic infection, or sepsis.
Patients should be closely monitored for the development of signs and symptoms of tuberculosis, including patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Routine monitoring of liver tests and prompt investigation of the causes of liver enzyme elevations is recommended to identify potential cases of drug-induced liver injury.
IV Compatibility
There is limited information regarding IV Compatibility of Tofacitinib in the drug label.
Overdosage
Signs, Symptoms, and Laboratory Findings of Acute Overdosage in Humans
There is no experience with overdose of XELJANZ.
Treatment or Management of Overdose
Pharmacokinetic data up to and including a single dose of 100 mg in healthy volunteers indicate that more than 95% of the administered dose is expected to be eliminated within 24 hours.
There is no specific antidote for overdose with XELJANZ. In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions. Patients who develop adverse reactions should receive appropriate treatment.
Pharmacology
There is limited information regarding Tofacitinib Pharmacology in the drug label.


{kind=link}