Teduglutide: Difference between revisions
No edit summary |
m (Protected "Teduglutide": Bot: Protecting all pages from category Drug ([Edit=Allow only administrators] (indefinite) [Move=Allow only administrators] (indefinite))) |
||
| (One intermediate revision by one other user not shown) | |||
| Line 1: | Line 1: | ||
{{DrugProjectFormSinglePage | {{DrugProjectFormSinglePage | ||
|authorTag= | |authorTag={{VP}}<!--Overview--> | ||
|aOrAn=a | |||
{{VP}} | |drugClass=[[glucagon-like peptide-2]] (GLP-2) analog | ||
|indicationType=treatment | |||
<!--Overview--> | |indication=[[short bowel syndrome]] (SBS) who are dependent on parenteral support | ||
|adverseReactions=[[abdominal pain]], [[injection site reaction]]s, [[nausea]], [[headaches]], [[abdominal distension]], [[upper respiratory tract infection]] | |||
|aOrAn= | |||
a | |||
|drugClass= | |||
[[glucagon-like peptide-2]] (GLP-2) analog | |||
|indication= | |||
[[short bowel syndrome]] (SBS) who are dependent on parenteral support | |||
|adverseReactions= | |||
[[abdominal pain]], [[injection site reaction]]s, [[nausea]], [[headaches]], [[abdominal distension]], [[upper respiratory tract infection]] | |||
<!--Black Box Warning--> | <!--Black Box Warning--> | ||
|blackBoxWarningTitle=Title | |||
|blackBoxWarningTitle= | |blackBoxWarningBody=<i><span style="color:#FF0000;">ConditionName: </span></i> | ||
Title | |||
|blackBoxWarningBody= | |||
<i><span style="color:#FF0000;">ConditionName: </span></i> | |||
* Content | * Content | ||
| Line 41: | Line 16: | ||
<!--FDA-Labeled Indications and Dosage (Adult)--> | <!--FDA-Labeled Indications and Dosage (Adult)--> | ||
|fdaLIADAdult======Short Bowel Syndrome===== | |||
|fdaLIADAdult= | |||
=====Short Bowel Syndrome===== | |||
*The recommended daily dose of GATTEX is 0.05 mg/kg body weight administered by subcutaneous injection once daily. Alternation of sites for [[subcutaneous]] injection is recommended, and can include the thighs, arms, and the quadrants of the abdomen. GATTEX should not be administered intravenously or [[intramuscularly]]. If a dose is missed, that dose should be taken as soon as possible on that day. Do not take 2 doses on the same day. | *The recommended daily dose of GATTEX is 0.05 mg/kg body weight administered by subcutaneous injection once daily. Alternation of sites for [[subcutaneous]] injection is recommended, and can include the thighs, arms, and the quadrants of the abdomen. GATTEX should not be administered intravenously or [[intramuscularly]]. If a dose is missed, that dose should be taken as soon as possible on that day. Do not take 2 doses on the same day. | ||
| Line 51: | Line 23: | ||
<!--Guideline-Supported Use (Adult)--> | <!--Guideline-Supported Use (Adult)--> | ||
|offLabelAdultGuideSupport=There is limited information regarding <i>Off-Label Guideline-Supported Use</i> of {{PAGENAME}} in adult patients. | |||
|offLabelAdultGuideSupport= | |||
There is limited information regarding <i>Off-Label Guideline-Supported Use</i> of {{PAGENAME}} in adult patients. | |||
<!--Non–Guideline-Supported Use (Adult)--> | <!--Non–Guideline-Supported Use (Adult)--> | ||
|offLabelAdultNoGuideSupport=There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in adult patients. | |||
|offLabelAdultNoGuideSupport= | |||
There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in adult patients. | |||
<!--Pediatric Indications and Dosage--> | <!--Pediatric Indications and Dosage--> | ||
<!--FDA-Labeled Indications and Dosage (Pediatric)--> | <!--FDA-Labeled Indications and Dosage (Pediatric)--> | ||
|fdaLIADPed=*Safety and efficacy in pediatric patients have not been established. | |||
|fdaLIADPed= | |||
*Safety and efficacy in pediatric patients have not been established. | |||
<!--Off-Label Use and Dosage (Pediatric)--> | <!--Off-Label Use and Dosage (Pediatric)--> | ||
<!--Guideline-Supported Use (Pediatric)--> | <!--Guideline-Supported Use (Pediatric)--> | ||
|offLabelPedGuideSupport=There is limited information regarding <i>Off-Label Guideline-Supported Use</i> of {{PAGENAME}} in pediatric patients. | |||
|offLabelPedGuideSupport= | |||
There is limited information regarding <i>Off-Label Guideline-Supported Use</i> of {{PAGENAME}} in pediatric patients. | |||
<!--Non–Guideline-Supported Use (Pediatric)--> | <!--Non–Guideline-Supported Use (Pediatric)--> | ||
|offLabelPedNoGuideSupport=There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in pediatric patients. | |||
|offLabelPedNoGuideSupport= | |||
There is limited information regarding <i>Off-Label Non–Guideline-Supported Use</i> of {{PAGENAME}} in pediatric patients. | |||
<!--Contraindications--> | <!--Contraindications--> | ||
|contraindications=* None. | |||
|contraindications= | |||
* None. | |||
<!--Warnings--> | <!--Warnings--> | ||
|warnings=====Precautions==== | |||
|warnings= | |||
====Precautions==== | |||
* Acceleration of Neoplastic Growth | * Acceleration of Neoplastic Growth | ||
| Line 122: | Line 73: | ||
<!--Clinical Trials Experience--> | <!--Clinical Trials Experience--> | ||
|clinicalTrials=*Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice. | |||
|clinicalTrials= | |||
*Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice. | |||
*Across all clinical studies, 566 subjects were exposed to at least one dose of GATTEX (190 patient-years of exposure; mean duration of exposure was 17 weeks). Of the 566 subjects, 173 subjects were treated in Phase 3 SBS studies (134/173 [77%] at the dose of 0.05 mg/kg/day and 39/173 [23%] at the dose of 0.10 mg/kg/day). | *Across all clinical studies, 566 subjects were exposed to at least one dose of GATTEX (190 patient-years of exposure; mean duration of exposure was 17 weeks). Of the 566 subjects, 173 subjects were treated in Phase 3 SBS studies (134/173 [77%] at the dose of 0.05 mg/kg/day and 39/173 [23%] at the dose of 0.10 mg/kg/day). | ||
| Line 152: | Line 100: | ||
<!--Postmarketing Experience--> | <!--Postmarketing Experience--> | ||
|postmarketing=There is limited information regarding <i>Postmarketing Experience</i> of {{PAGENAME}} in the drug label. | |||
|postmarketing= | |||
There is limited information regarding <i>Postmarketing Experience</i> of {{PAGENAME}} in the drug label. | |||
<!--Drug Interactions--> | <!--Drug Interactions--> | ||
|drugInteractions=* Potential for Increased Absorption of Oral Medications | |||
|drugInteractions= | |||
* Potential for Increased Absorption of Oral Medications | |||
:*Based upon the pharmacodynamic effect of GATTEX, there is a potential for increased absorption of concomitant oral medications, which should be considered if these drugs require titration or have a narrow therapeutic index. | :*Based upon the pharmacodynamic effect of GATTEX, there is a potential for increased absorption of concomitant oral medications, which should be considered if these drugs require titration or have a narrow therapeutic index. | ||
| Line 168: | Line 110: | ||
<!--Use in Specific Populations--> | <!--Use in Specific Populations--> | ||
|useInPregnancyFDA=* '''Pregnancy Category B''' | |||
|useInPregnancyFDA= | |||
* '''Pregnancy Category B''' | |||
*Reproduction studies with teduglutide have been performed in pregnant rats at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg) and in rabbits at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg). These studies did not reveal any evidence of impaired fertility or harm to the fetus due to teduglutide. A pre- and postnatal development study in rats showed no evidence of any adverse effect on pre- and postnatal development at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg). There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, teduglutide should be used during pregnancy only if clearly needed. | *Reproduction studies with teduglutide have been performed in pregnant rats at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg) and in rabbits at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg). These studies did not reveal any evidence of impaired fertility or harm to the fetus due to teduglutide. A pre- and postnatal development study in rats showed no evidence of any adverse effect on pre- and postnatal development at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg). There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, teduglutide should be used during pregnancy only if clearly needed. | ||
|useInPregnancyAUS=* '''Australian Drug Evaluation Committee (ADEC) Pregnancy Category''' | |||
|useInPregnancyAUS= | |||
* '''Australian Drug Evaluation Committee (ADEC) Pregnancy Category''' | |||
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of {{PAGENAME}} in women who are pregnant. | There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of {{PAGENAME}} in women who are pregnant. | ||
|useInLaborDelivery=There is no FDA guidance on use of {{PAGENAME}} during labor and delivery. | |||
|useInLaborDelivery= | |useInNursing=*It is unknown whether teduglutide is excreted in human milk. Teduglutide is excreted in the milk of lactating rats, and the highest concentration in the milk was 2.9% of the plasma concentration following a single subcutaneous injection of 25 mg/kg. Because many drugs are excreted in human milk; because of the potential for serious adverse reactions to nursing infants from teduglutide and because of the potential for tumorigenicity shown for teduglutide in rats, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. | ||
There is no FDA guidance on use of {{PAGENAME}} during labor and delivery. | |useInPed=*Safety and efficacy in pediatric patients have not been established. | ||
|useInGeri=*No dose adjustment is necessary in patients above the age of 65 years. Of the 566 subjects treated with teduglutide, 43 subjects were 65 years or older, whereas 6 subjects were 75 years of age or older. In the SBS studies, no overall differences in safety or efficacy were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. | |||
|useInNursing= | |useInGender=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific gender populations. | ||
|useInRace=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific racial populations. | |||
*It is unknown whether teduglutide is excreted in human milk. Teduglutide is excreted in the milk of lactating rats, and the highest concentration in the milk was 2.9% of the plasma concentration following a single subcutaneous injection of 25 mg/kg. Because many drugs are excreted in human milk; because of the potential for serious adverse reactions to nursing infants from teduglutide and because of the potential for tumorigenicity shown for teduglutide in rats, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. | |useInRenalImpair=*Reduce the dose of GATTEX by 50% in patients with moderate and severe [[renal impairment]] ([[creatinine clearance]] less than 50 mL/min) and [[end-stage renal disease]] (ESRD). | ||
|useInHepaticImpair=*GATTEX has not been formally studied in subjects with severe [[hepatic impairment]]. No dosage adjustment is necessary for patients with mild and moderate [[hepatic impairment]] based on a study conducted in Child-Pugh grade B subjects. | |||
|useInPed= | |useInReproPotential=There is no FDA guidance on the use of {{PAGENAME}} in women of reproductive potentials and males. | ||
|useInImmunocomp=There is no FDA guidance one the use of {{PAGENAME}} in patients who are immunocompromised. | |||
*Safety and efficacy in pediatric patients have not been established. | |||
|useInGeri= | |||
*No dose adjustment is necessary in patients above the age of 65 years. Of the 566 subjects treated with teduglutide, 43 subjects were 65 years or older, whereas 6 subjects were 75 years of age or older. In the SBS studies, no overall differences in safety or efficacy were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. | |||
|useInGender= | |||
There is no FDA guidance on the use of {{PAGENAME}} with respect to specific gender populations. | |||
|useInRace= | |||
There is no FDA guidance on the use of {{PAGENAME}} with respect to specific racial populations. | |||
|useInRenalImpair= | |||
*Reduce the dose of GATTEX by 50% in patients with moderate and severe [[renal impairment]] ([[creatinine clearance]] less than 50 mL/min) and [[end-stage renal disease]] (ESRD). | |||
|useInHepaticImpair= | |||
*GATTEX has not been formally studied in subjects with severe [[hepatic impairment]]. No dosage adjustment is necessary for patients with mild and moderate [[hepatic impairment]] based on a study conducted in Child-Pugh grade B subjects. | |||
|useInReproPotential= | |||
There is no FDA guidance on the use of {{PAGENAME}} in women of reproductive potentials and males. | |||
|useInImmunocomp= | |||
There is no FDA guidance one the use of {{PAGENAME}} in patients who are immunocompromised. | |||
<!--Administration and Monitoring--> | <!--Administration and Monitoring--> | ||
|administration=* Subcutaneous | |||
|administration= | |monitoring=There is limited information regarding <i>Monitoring</i> of {{PAGENAME}} in the drug label. | ||
* Subcutaneous | |||
|monitoring= | |||
There is limited information regarding <i>Monitoring</i> of {{PAGENAME}} in the drug label. | |||
<!--IV Compatibility--> | <!--IV Compatibility--> | ||
|IVCompat=There is limited information regarding <i>IV Compatibility</i> of {{PAGENAME}} in the drug label. | |||
|IVCompat= | |||
There is limited information regarding <i>IV Compatibility</i> of {{PAGENAME}} in the drug label. | |||
<!--Overdosage--> | <!--Overdosage--> | ||
|overdose====Acute Overdose=== | |||
|overdose= | |||
===Acute Overdose=== | |||
*The maximum dose of GATTEX studied during clinical development was 80 mg/day for 8 days. In the event of overdose, the patient should be carefully monitored by the medical professional. | *The maximum dose of GATTEX studied during clinical development was 80 mg/day for 8 days. In the event of overdose, the patient should be carefully monitored by the medical professional. | ||
| Line 245: | Line 146: | ||
<!--Drug box 2--> | <!--Drug box 2--> | ||
|drugBox={{Drugbox2 | |||
|drugBox= | |||
{{Drugbox2 | |||
| Watchedfields = changed | | Watchedfields = changed | ||
| verifiedrevid = 443663022 | | verifiedrevid = 443663022 | ||
| Line 307: | Line 205: | ||
<!--Mechanism of Action--> | <!--Mechanism of Action--> | ||
|mechAction=* Teduglutide is an analog of naturally occurring human [[glucagon-like peptide-2]] (GLP-2), a peptide secreted by L-cells of the distal intestine. GLP-2 is known to increase intestinal and portal blood flow, and inhibit gastric acid secretion. Teduglutide binds to the [[glucagon-like peptide-2]] receptors located in intestinal subpopulations of enteroendocrine cells, subepithelial [[myofibroblasts]] and enteric neurons of the submucosal and [[myenteric plexus]]. Activation of these receptors results in the local release of multiple mediators including [[insulin-like growth factor]] (IGF)-1, [[nitric oxide]] and [[keratinocyte]] growth factor (KGF). | |||
|mechAction= | |||
* Teduglutide is an analog of naturally occurring human [[glucagon-like peptide-2]] (GLP-2), a peptide secreted by L-cells of the distal intestine. GLP-2 is known to increase intestinal and portal blood flow, and inhibit gastric acid secretion. Teduglutide binds to the [[glucagon-like peptide-2]] receptors located in intestinal subpopulations of enteroendocrine cells, subepithelial [[myofibroblasts]] and enteric neurons of the submucosal and [[myenteric plexus]]. Activation of these receptors results in the local release of multiple mediators including [[insulin-like growth factor]] (IGF)-1, [[nitric oxide]] and [[keratinocyte]] growth factor (KGF). | |||
<!--Structure--> | <!--Structure--> | ||
|structure=* The active ingredient in GATTEX (teduglutide [rDNA origin]) for injection is teduglutide (rDNA origin), which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified by recombinant DNA technology. The chemical name of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is: | |||
|structure= | |||
* The active ingredient in GATTEX (teduglutide [rDNA origin]) for injection is teduglutide (rDNA origin), which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified by recombinant DNA technology. The chemical name of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is: | |||
: [[File:{{PAGENAME}}08.png|thumb|none|600px|This image is provided by the National Library of Medicine.]] | : [[File:{{PAGENAME}}08.png|thumb|none|600px|This image is provided by the National Library of Medicine.]] | ||
| Line 327: | Line 219: | ||
<!--Pharmacodynamics--> | <!--Pharmacodynamics--> | ||
|PD=*The ability of GATTEX to improve intestinal absorption was studied in 17 adult subjects with Short Bowel Syndrome using daily doses of 0.03, 0.10, 0.15 mg/kg (N=2-3 per dose group) in a 21-day, open-label, multi-center, dose-ranging study. All subcutaneous (abdomen) doses studied, except 0.03 mg/kg once daily, resulted in enhanced gastrointestinal fluid (wet weight) absorption of approximately 750-1000 mL/day, and increased villus height and crypt depth of the intestinal mucosa. | |||
|PD= | |||
*The ability of GATTEX to improve intestinal absorption was studied in 17 adult subjects with Short Bowel Syndrome using daily doses of 0.03, 0.10, 0.15 mg/kg (N=2-3 per dose group) in a 21-day, open-label, multi-center, dose-ranging study. All subcutaneous (abdomen) doses studied, except 0.03 mg/kg once daily, resulted in enhanced gastrointestinal fluid (wet weight) absorption of approximately 750-1000 mL/day, and increased villus height and crypt depth of the intestinal mucosa. | |||
*At a dose 5 times the maximum recommended dose, teduglutide did not prolong the [[QTc interval]] to any clinically relevant extent. | *At a dose 5 times the maximum recommended dose, teduglutide did not prolong the [[QTc interval]] to any clinically relevant extent. | ||
<!--Pharmacokinetics--> | <!--Pharmacokinetics--> | ||
|PK=*Absorption: | |||
|PK= | |||
*Absorption: | |||
:*In healthy subjects, GATTEX administered subcutaneously had an absolute bioavailability of 88% and reached maximum plasma teduglutide concentrations at 3-5 hours after administration. Following a 0.05 mg/kg subcutaneous dose in SBS subjects, the median peak teduglutide concentration (Cmax) was 36 ng/mL and the median area under the curve (AUC0-inf) was 0.15 µg•hr/mL. No accumulation of teduglutide was observed following repeated subcutaneous administrations. | :*In healthy subjects, GATTEX administered subcutaneously had an absolute bioavailability of 88% and reached maximum plasma teduglutide concentrations at 3-5 hours after administration. Following a 0.05 mg/kg subcutaneous dose in SBS subjects, the median peak teduglutide concentration (Cmax) was 36 ng/mL and the median area under the curve (AUC0-inf) was 0.15 µg•hr/mL. No accumulation of teduglutide was observed following repeated subcutaneous administrations. | ||
| Line 366: | Line 252: | ||
<!--Nonclinical Toxicology--> | <!--Nonclinical Toxicology--> | ||
|nonClinToxic=*In a 2-year carcinogenicity study in Wistar Han rats at subcutaneous doses of 3, 10 and 35 mg/kg/day (about 60, 200 and 700 times the recommended daily human dose of 0.05 mg/kg, respectively), teduglutide caused statistically significant increases in the incidences of adenomas in the bile duct and jejunum of male rats. | |||
|nonClinToxic= | |||
*In a 2-year carcinogenicity study in Wistar Han rats at subcutaneous doses of 3, 10 and 35 mg/kg/day (about 60, 200 and 700 times the recommended daily human dose of 0.05 mg/kg, respectively), teduglutide caused statistically significant increases in the incidences of adenomas in the bile duct and jejunum of male rats. | |||
*Teduglutide was negative in the Ames test, chromosomal aberration test in Chinese hamster ovary cells, and in vivo mouse micronucleus assay. | *Teduglutide was negative in the Ames test, chromosomal aberration test in Chinese hamster ovary cells, and in vivo mouse micronucleus assay. | ||
| Line 376: | Line 259: | ||
<!--Clinical Studies--> | <!--Clinical Studies--> | ||
|clinicalStudies======Study 1 (Placebo-controlled) and Study 2 (Open-label extension of Study 1)===== | |||
|clinicalStudies= | |||
=====Study 1 (Placebo-controlled) and Study 2 (Open-label extension of Study 1)===== | |||
*Study 1. The efficacy, safety, and tolerability of GATTEX was evaluated in a randomized, double-blind, placebo-controlled, parallel-group, multi-national, multi-center clinical trial (Study 1) in adults with SBS who were dependent on [[parenteral nutrition]]/intravenous (PN/I.V.) support for at least 12 months and required PN at least 3 times per week. For 8 weeks (or less) prior to randomization, investigators optimized the PN/I.V. volume of all subjects. Optimization was followed by a 4-week to 8-week period of fluid stabilization. Subjects then were randomized (1:1) to placebo (n=43) or GATTEX 0.05 mg/kg/day (n=43). Study treatment was administered subcutaneously once daily for 24 weeks. PN/I.V. volume adjustments (up to 30% decrease) and clinical assessments were made at 2, 4, 8, 12, 20, and 24 weeks. | *Study 1. The efficacy, safety, and tolerability of GATTEX was evaluated in a randomized, double-blind, placebo-controlled, parallel-group, multi-national, multi-center clinical trial (Study 1) in adults with SBS who were dependent on [[parenteral nutrition]]/intravenous (PN/I.V.) support for at least 12 months and required PN at least 3 times per week. For 8 weeks (or less) prior to randomization, investigators optimized the PN/I.V. volume of all subjects. Optimization was followed by a 4-week to 8-week period of fluid stabilization. Subjects then were randomized (1:1) to placebo (n=43) or GATTEX 0.05 mg/kg/day (n=43). Study treatment was administered subcutaneously once daily for 24 weeks. PN/I.V. volume adjustments (up to 30% decrease) and clinical assessments were made at 2, 4, 8, 12, 20, and 24 weeks. | ||
| Line 406: | Line 286: | ||
<!--How Supplied--> | <!--How Supplied--> | ||
|howSupplied=* GATTEX® (teduglutide [rDNA origin]) for injection is supplied in a sterile, single-use glass vial containing 5 mg of teduglutide as a white, lyophilized powder to be reconstituted with 0.5 mL Sterile Water for Injection. The product to be dispensed is either a one-vial kit or a 30-vial kit. The one-vial kit is pre-assembled and ready to be used. The 30-vial kit is to be assembled by a pharmacist with the following two cartons: | |||
|howSupplied= | |||
* GATTEX® (teduglutide [rDNA origin]) for injection is supplied in a sterile, single-use glass vial containing 5 mg of teduglutide as a white, lyophilized powder to be reconstituted with 0.5 mL Sterile Water for Injection. The product to be dispensed is either a one-vial kit or a 30-vial kit. The one-vial kit is pre-assembled and ready to be used. The 30-vial kit is to be assembled by a pharmacist with the following two cartons: | |||
*Carton of Drug Vials (NDC 68875-0101-2): | *Carton of Drug Vials (NDC 68875-0101-2): | ||
| Line 450: | Line 327: | ||
<!--Patient Counseling Information--> | <!--Patient Counseling Information--> | ||
|fdaPatientInfo=*General Counseling Information | |||
|fdaPatientInfo= | |||
*General Counseling Information | |||
:*Prior to treatment, patients should fully understand the risks and benefits of GATTEX. Ensure that all patients receive the Medication Guide prior to initiating GATTEX therapy. | :*Prior to treatment, patients should fully understand the risks and benefits of GATTEX. Ensure that all patients receive the Medication Guide prior to initiating GATTEX therapy. | ||
| Line 488: | Line 362: | ||
<!--Precautions with Alcohol--> | <!--Precautions with Alcohol--> | ||
|alcohol=* Alcohol-{{PAGENAME}} interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication. | |||
|alcohol= | |||
* Alcohol-{{PAGENAME}} interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication. | |||
<!--Brand Names--> | <!--Brand Names--> | ||
|brandNames=* GATTEX®<ref>{{Cite web | title = GATTEX teduglutide injection, powder, for solution | url = http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=66b69c1e-b25c-44d3-b5ff-1c1de9a516fa }}</ref> | |||
|brandNames= | |||
* GATTEX®<ref>{{Cite web | title = GATTEX teduglutide injection, powder, for solution | url = http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=66b69c1e-b25c-44d3-b5ff-1c1de9a516fa }}</ref> | |||
<!--Look-Alike Drug Names--> | <!--Look-Alike Drug Names--> | ||
|lookAlike=<!--Drug Shortage Status--> | |||
|lookAlike= | |||
<!--Drug Shortage Status--> | |||
|drugShortage= | |drugShortage= | ||
}} | }} | ||
{{PillImage | {{PillImage | ||
|fileName=No image.jpg | |fileName=No image.jpg | ||
}} | }} | ||
{{LabelImage | {{LabelImage | ||
|fileName={{PAGENAME}}03.png | |fileName={{PAGENAME}}03.png | ||
}} | }} | ||
{{LabelImage | {{LabelImage | ||
|fileName={{PAGENAME}}04.png | |fileName={{PAGENAME}}04.png | ||
}} | }} | ||
{{LabelImage | {{LabelImage | ||
|fileName={{PAGENAME}}05.png | |fileName={{PAGENAME}}05.png | ||
}} | }} | ||
{{LabelImage | {{LabelImage | ||
|fileName={{PAGENAME}}06.png | |fileName={{PAGENAME}}06.png | ||
}} | }} | ||
<!--Pill Image--> | |||
<!--Label Display Image--> | |||
<!--Category--> | <!--Category--> | ||
[[Category:Drug]] | [[Category:Drug]] | ||
Latest revision as of 17:13, 20 August 2015
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Vignesh Ponnusamy, M.B.B.S. [2]
Disclaimer
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Teduglutide is a glucagon-like peptide-2 (GLP-2) analog that is FDA approved for the treatment of short bowel syndrome (SBS) who are dependent on parenteral support. Common adverse reactions include abdominal pain, injection site reactions, nausea, headaches, abdominal distension, upper respiratory tract infection.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Short Bowel Syndrome
- The recommended daily dose of GATTEX is 0.05 mg/kg body weight administered by subcutaneous injection once daily. Alternation of sites for subcutaneous injection is recommended, and can include the thighs, arms, and the quadrants of the abdomen. GATTEX should not be administered intravenously or intramuscularly. If a dose is missed, that dose should be taken as soon as possible on that day. Do not take 2 doses on the same day.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Teduglutide in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Teduglutide in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
- Safety and efficacy in pediatric patients have not been established.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Teduglutide in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Teduglutide in pediatric patients.
Contraindications
- None.
Warnings
Precautions
- Acceleration of Neoplastic Growth
- Based on the pharmacologic activity and findings in animals, GATTEX has the potential to cause hyperplastic changes including neoplasia. In patients at increased risk for malignancy, the clinical decision to use GATTEX should be considered only if the benefits outweigh the risks. In patients with active gastrointestinal malignancy (GI tract, hepatobiliary, pancreatic), GATTEX therapy should be discontinued. In patients with active non-gastrointestinal malignancy, the clinical decision to continue GATTEX should be made based on risk-benefit considerations.
- Colorectal Polyps
- Colorectal polyps were identified during the clinical trials. Colonoscopy of the entire colon with removal of polyps should be done within 6 months prior to starting treatment with GATTEX. A follow-up colonoscopy (or alternate imaging) is recommended at the end of 1 year of GATTEX. Subsequent colonoscopies should be done every 5 years or more often as needed. If a polyp is found, adherence to current polyp follow-up guidelines is recommended. In case of diagnosis of colorectal cancer, GATTEX therapy should be discontinued.
- Small Bowel Neoplasia
- Based on benign tumor findings in the rat carcinogenicity study, patients should be monitored clinically for small bowel neoplasia. If a benign neoplasm is found, it should be removed. In case of small bowel cancer, GATTEX therapy should be discontinued.
- Intestinal Obstruction
- Intestinal obstruction has been reported in clinical trials. In patients who develop intestinal or stomal obstruction, GATTEX should be temporarily discontinued while the patient is clinically managed. GATTEX may be restarted when the obstructive presentation resolves, if clinically indicated.
- Biliary and Pancreatic Disease
- Gallbladder and Biliary Tract Disease
- Cholecystitis, cholangitis, and cholelithiasis, have been reported in clinical studies. For identification of the onset or worsening of gallbladder/biliary disease, patients should undergo laboratory assessment of bilirubin and alkaline phosphatase within 6 months prior to starting GATTEX, and at least every 6 months while on GATTEX; or more frequently if needed. If clinically meaningful changes are seen, further evaluation including imaging of the gallbladder and/or biliary tract is recommended; and the need for continued GATTEX treatment should be reassessed.
- Pancreatic Disease
- Pancreatitis has been reported in clinical studies. For identification of onset or worsening of pancreatic disease, patients should undergo laboratory assessment of lipase and amylase within 6 months prior to starting GATTEX, and at least every 6 months while on GATTEX; or more frequently if needed. If clinically meaningful changes are seen, further evaluation such as imaging of the pancreas is recommended; and the need for continued GATTEX treatment should be reassessed.
- Fluid Overload
- Fluid overload and congestive heart failure have been observed in clinical trials, which were felt to be related to enhanced fluid absorption associated with GATTEX. If fluid overload occurs, parenteral support should be adjusted and GATTEX treatment should be reassessed, especially in patients with underlying cardiovascular disease. If significant cardiac deterioration develops while on GATTEX, the need for continued GATTEX treatment should be reassessed.
- Increased Absorption of Concomitant Oral Medication
- Altered mental status in association with GATTEX has been observed in patients on benzodiazepines in clinical trials. Patients on concomitant oral drugs (e.g., benzodiazepines, phenothiazines) requiring titration or with a narrow therapeutic index may require dose adjustment while on GATTEX.
Adverse Reactions
Clinical Trials Experience
- Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
- Across all clinical studies, 566 subjects were exposed to at least one dose of GATTEX (190 patient-years of exposure; mean duration of exposure was 17 weeks). Of the 566 subjects, 173 subjects were treated in Phase 3 SBS studies (134/173 [77%] at the dose of 0.05 mg/kg/day and 39/173 [23%] at the dose of 0.10 mg/kg/day).
- The most commonly reported (≥ 10%) adverse reactions in patients treated with GATTEX across all clinical studies (n = 566) were: abdominal pain (30.0%); injection site reactions (22.4%); nausea (18.2%); headaches (15.9%); abdominal distension (13.8%); upper respiratory tract infection (11.8%).
- The rates of adverse reactions in subjects with SBS participating in two randomized, placebo-controlled, 24-week, double-blind clinical studies (Study 1 and Study 3) are summarized in Table 1. Only those reactions with a rate of at least 5% in the GATTEX group, and greater than placebo group, are summarized in Table 1. The majority of these reactions were mild or moderate. Of subjects receiving GATTEX at the recommended dose of 0.05 mg/kg/day, 88.3% (N=68/77) experienced an adverse reaction, as compared to 83.1% (49/59) for placebo. Many of these adverse reactions have been reported in association with the underlying disease and/or parenteral nutrition.
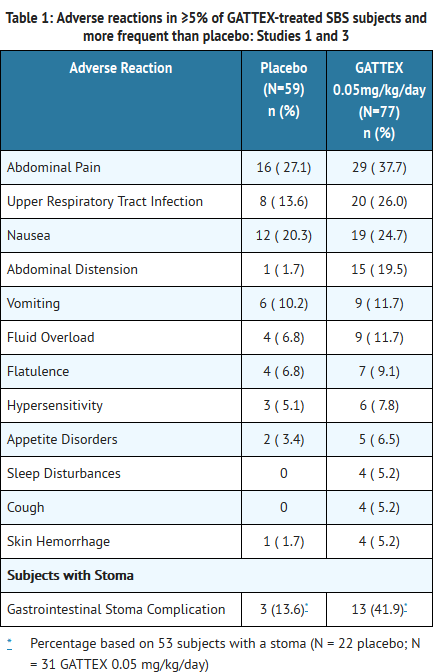
This image is provided by the National Library of Medicine.

- In placebo-controlled Studies 1 and 3, 12% of patients in each of the placebo and GATTEX study groups experienced an injection site reaction.
Adverse Reactions of Special Interest
- Malignancy. Three subjects were diagnosed with malignancy in the clinical studies, all of whom were male and had received GATTEX 0.05 mg/kg/day in Study 2. One subject had a history of abdominal radiation for Hodgkin's disease two decades prior to receiving GATTEX and prior liver lesion on CT scan, and was diagnosed with metastatic adenocarcinoma of unconfirmed origin after 11 months of exposure to GATTEX. Two subjects had extensive smoking histories, and were diagnosed with lung cancers (squamous and non-small cell) after 12 months and 3 months of GATTEX exposure, respectively.
- Colorectal Polyps. In the clinical studies, 6 subjects were diagnosed with polyps of the G.I. tract after initiation of study treatment. In the SBS placebo-controlled studies, 1/59 (1.7%) of subjects on placebo and 1/109 (0.9%) of subjects on GATTEX 0.05 mg/kg/day were diagnosed with intestinal polyps (inflammatory stomal and hyperplastic sigmoidal after 3 and 5 months, respectively). The remaining 4 polyp cases occurred in the extension studies – two colorectal villous adenomas (onset at 6 and 7 months in GATTEX 0.10 and 0.05 mg/kg/day dose groups, respectively), one hyperplastic polyp (onset 6 months in GATTEX 0.10 mg/kg/day dose group), and one small duodenal polyp (onset at 3 months in GATTEX 0.05 mg/kg/day dose group).
- Gastrointestinal Obstruction. Overall, 12 subjects experienced one or more episodes of intestinal obstruction/stenosis: 6 in SBS placebo-controlled studies and 6 in the extension studies. The 6 subjects in the placebo-controlled trials were all on GATTEX: 3/77 (3.9%) on GATTEX 0.05 mg/kg/day and 3/32 (9.4%) on GATTEX 0.10 mg/kg/day. No cases of intestinal obstruction occurred in the placebo group. Onsets ranged from 1 day to 6 months. In the extension studies, 6 additional subjects (all on GATTEX 0.05 mg/kg/day) were diagnosed with intestinal obstruction/stenosis with onsets ranging from 6 days to 7 months. Two of the 6 subjects from the placebo-controlled trials experienced recurrence of obstruction in the extension studies. Of all 8 subjects with an episode of intestinal obstruction/stenosis in these extension studies, 1 subject required endoscopic dilation and none required surgical intervention.
- Gallbladder, Biliary and Pancreatic Disease. For gallbladder and biliary disease in the placebo-controlled studies, 3 subjects were diagnosed with cholecystitis, all of whom had a prior history of gallbladder disease and were in the GATTEX 0.05 mg/kg/day dose group. No cases were reported in the placebo group. One of these 3 cases had gallbladder perforation and underwent cholecystectomy the next day. The remaining 2 cases underwent elective cholecystectomy at a later date. In the extension studies, 3 subjects had an episode of acute cholecystitis; 2 subjects had new-onset cholelithiasis; and 1 subject experienced cholestasis secondary to an obstructed biliary stent. For pancreatic disease in the placebo-controlled studies, 1 subject (GATTEX 0.05 mg/kg/day dose group) had a pancreatic pseudocyst diagnosed after 4 months of GATTEX. In the extension studies, 1 subject was diagnosed with chronic pancreatitis; and 1 subject was diagnosed with acute pancreatitis.
- Fluid Overload. In the placebo-controlled trials, fluid overload was reported in 4/59 (6.8%) of subjects on placebo and 9/77 (11.7%) subjects on GATTEX 0.05 mg/kg/day. Of the 9 cases in the GATTEX group, there were 2 cases of congestive heart failure (CHF), 1 of whom was reported as a serious adverse event and the other as non-serious. The serious case had onset at 6 months, and was possibly associated with previously undiagnosed hypothyroidism and/or cardiac dysfunction.
- Concomitant Oral Medication. GATTEX can increase the absorption of concomitant oral medications such as benzodiazepines and psychotropic agents. In the placebo-controlled trials, an analysis of episodes of cognition and attention disturbances was performed for subjects on benzodiazepines. One of the subjects in the GATTEX 0.05 mg/kg/day group (on prazepam) experienced dramatic deterioration in mental status progressing to coma during her first week of GATTEX therapy. She was admitted to the ICU where her benzodiazepine level was >300 mcg/L. GATTEX and prazepam were discontinued, and coma resolved 5 days later.
Postmarketing Experience
There is limited information regarding Postmarketing Experience of Teduglutide in the drug label.
Drug Interactions
- Potential for Increased Absorption of Oral Medications
- Based upon the pharmacodynamic effect of GATTEX, there is a potential for increased absorption of concomitant oral medications, which should be considered if these drugs require titration or have a narrow therapeutic index.
- Concomitant Drug Therapy
- Clinical interaction studies were not performed. No inhibition or induction of the cytochrome P450 enzyme system has been observed based on in vitro studies although the relevance of in vitro studies to an in vivo setting is unknown.
Use in Specific Populations
Pregnancy
- Pregnancy Category B
- Reproduction studies with teduglutide have been performed in pregnant rats at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg) and in rabbits at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg). These studies did not reveal any evidence of impaired fertility or harm to the fetus due to teduglutide. A pre- and postnatal development study in rats showed no evidence of any adverse effect on pre- and postnatal development at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg). There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, teduglutide should be used during pregnancy only if clearly needed.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Teduglutide in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Teduglutide during labor and delivery.
Nursing Mothers
- It is unknown whether teduglutide is excreted in human milk. Teduglutide is excreted in the milk of lactating rats, and the highest concentration in the milk was 2.9% of the plasma concentration following a single subcutaneous injection of 25 mg/kg. Because many drugs are excreted in human milk; because of the potential for serious adverse reactions to nursing infants from teduglutide and because of the potential for tumorigenicity shown for teduglutide in rats, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
- Safety and efficacy in pediatric patients have not been established.
Geriatic Use
- No dose adjustment is necessary in patients above the age of 65 years. Of the 566 subjects treated with teduglutide, 43 subjects were 65 years or older, whereas 6 subjects were 75 years of age or older. In the SBS studies, no overall differences in safety or efficacy were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Gender
There is no FDA guidance on the use of Teduglutide with respect to specific gender populations.
Race
There is no FDA guidance on the use of Teduglutide with respect to specific racial populations.
Renal Impairment
- Reduce the dose of GATTEX by 50% in patients with moderate and severe renal impairment (creatinine clearance less than 50 mL/min) and end-stage renal disease (ESRD).
Hepatic Impairment
- GATTEX has not been formally studied in subjects with severe hepatic impairment. No dosage adjustment is necessary for patients with mild and moderate hepatic impairment based on a study conducted in Child-Pugh grade B subjects.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Teduglutide in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Teduglutide in patients who are immunocompromised.
Administration and Monitoring
Administration
- Subcutaneous
Monitoring
There is limited information regarding Monitoring of Teduglutide in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Teduglutide in the drug label.
Overdosage
Acute Overdose
- The maximum dose of GATTEX studied during clinical development was 80 mg/day for 8 days. In the event of overdose, the patient should be carefully monitored by the medical professional.
Chronic Overdose
There is limited information regarding Chronic Overdose of Teduglutide in the drug label.
Pharmacology

Mechanism of Action
- Teduglutide is an analog of naturally occurring human glucagon-like peptide-2 (GLP-2), a peptide secreted by L-cells of the distal intestine. GLP-2 is known to increase intestinal and portal blood flow, and inhibit gastric acid secretion. Teduglutide binds to the glucagon-like peptide-2 receptors located in intestinal subpopulations of enteroendocrine cells, subepithelial myofibroblasts and enteric neurons of the submucosal and myenteric plexus. Activation of these receptors results in the local release of multiple mediators including insulin-like growth factor (IGF)-1, nitric oxide and keratinocyte growth factor (KGF).
Structure
- The active ingredient in GATTEX (teduglutide [rDNA origin]) for injection is teduglutide (rDNA origin), which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified by recombinant DNA technology. The chemical name of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is:
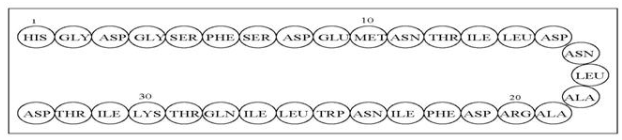
This image is provided by the National Library of Medicine.

- Teduglutide has a molecular weight of 3752 Daltons. Teduglutide drug substance is a clear, colorless to light-straw–colored liquid.
- Each single-use vial of GATTEX contains 5 mg of teduglutide as a white lyophilized powder for solution for subcutaneous injection. In addition to the active pharmaceutical ingredient (teduglutide), each vial of GATTEX contains 3.88 mg L-histidine, 15 mg mannitol, 0.644 mg monobasic sodium phosphate monohydrate, 3.434 mg dibasic sodium phosphate heptahydrate as excipients. No preservatives are present.
- At the time of administration the lyophilized powder is reconstituted with 0.5 mL of Sterile Water for Injection, which is provided in a prefilled syringe. A 10 mg/mL sterile solution is obtained after reconstitution. Up to 0.38 mL of the reconstituted solution which contains 3.8 mg of teduglutide can be withdrawn for subcutaneous injection upon reconstitution.
Pharmacodynamics
- The ability of GATTEX to improve intestinal absorption was studied in 17 adult subjects with Short Bowel Syndrome using daily doses of 0.03, 0.10, 0.15 mg/kg (N=2-3 per dose group) in a 21-day, open-label, multi-center, dose-ranging study. All subcutaneous (abdomen) doses studied, except 0.03 mg/kg once daily, resulted in enhanced gastrointestinal fluid (wet weight) absorption of approximately 750-1000 mL/day, and increased villus height and crypt depth of the intestinal mucosa.
- At a dose 5 times the maximum recommended dose, teduglutide did not prolong the QTc interval to any clinically relevant extent.
Pharmacokinetics
- Absorption:
- In healthy subjects, GATTEX administered subcutaneously had an absolute bioavailability of 88% and reached maximum plasma teduglutide concentrations at 3-5 hours after administration. Following a 0.05 mg/kg subcutaneous dose in SBS subjects, the median peak teduglutide concentration (Cmax) was 36 ng/mL and the median area under the curve (AUC0-inf) was 0.15 µg•hr/mL. No accumulation of teduglutide was observed following repeated subcutaneous administrations.
- Distribution:
- In healthy subjects, teduglutide has a volume of distribution (103 mL/kg) similar to blood volume.
- Metabolism:
- The metabolic pathway of teduglutide was not investigated in humans. However, teduglutide is expected to be degraded into small peptides and amino acids via catabolic pathways, similar to the catabolism of endogenous GLP-2.
- Elimination:
- In healthy subjects, teduglutide plasma clearance was approximately 123 mL/hr/kg which is similar to the GFR suggesting that teduglutide is primarily cleared by the kidney. Teduglutide has a mean terminal half-life (t1/2) of approximately 2 hours in healthy subjects and 1.3 hours in SBS subjects.
- Dose Linearity:
- The Cmax and AUC of teduglutide was dose proportional over the dose range of 0.05 to 0.4 mg/kg GATTEX.
- Hepatic Impairment:
- Subjects with moderate hepatic impairment had lower teduglutide Cmax and AUC (10 ~15%) compared to healthy matched control subjects after a single subcutaneous dose of 20 mg GATTEX. Teduglutide PK was not assessed in subjects with severe hepatic impairment.
- Renal Impairment:
- In subjects with moderate to severe renal impairment or end stage renal disease (ESRD), teduglutide Cmax and AUC0-inf increased with the degree of renal impairment following a single subcutaneous administration of 10 mg teduglutide. Teduglutide exposure increased by a factor of 2.1 (Cmax) and 2.6 (AUC0-inf) in ESRD subjects compared to healthy subjects.
- Geriatric Patients:
- No differences were observed between healthy subjects younger than 65 years and those older than 65 years. Experience in subjects 75 years and above is limited.
- Gender:
- No clinically relevant gender differences were observed.
Nonclinical Toxicology
- In a 2-year carcinogenicity study in Wistar Han rats at subcutaneous doses of 3, 10 and 35 mg/kg/day (about 60, 200 and 700 times the recommended daily human dose of 0.05 mg/kg, respectively), teduglutide caused statistically significant increases in the incidences of adenomas in the bile duct and jejunum of male rats.
- Teduglutide was negative in the Ames test, chromosomal aberration test in Chinese hamster ovary cells, and in vivo mouse micronucleus assay.
- Teduglutide at subcutaneous doses up to 50 mg/kg/day (about 1000 times the recommended daily human dose of 0.05 mg/kg) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
Clinical Studies
Study 1 (Placebo-controlled) and Study 2 (Open-label extension of Study 1)
- Study 1. The efficacy, safety, and tolerability of GATTEX was evaluated in a randomized, double-blind, placebo-controlled, parallel-group, multi-national, multi-center clinical trial (Study 1) in adults with SBS who were dependent on parenteral nutrition/intravenous (PN/I.V.) support for at least 12 months and required PN at least 3 times per week. For 8 weeks (or less) prior to randomization, investigators optimized the PN/I.V. volume of all subjects. Optimization was followed by a 4-week to 8-week period of fluid stabilization. Subjects then were randomized (1:1) to placebo (n=43) or GATTEX 0.05 mg/kg/day (n=43). Study treatment was administered subcutaneously once daily for 24 weeks. PN/I.V. volume adjustments (up to 30% decrease) and clinical assessments were made at 2, 4, 8, 12, 20, and 24 weeks.
- The primary efficacy endpoint was based on a clinical response, defined as a subject achieving at least 20% reduction in weekly PN/I.V. volume from Baseline (immediately before randomization) to both Weeks 20 and 24.
- The mean age of subjects was 50.3 years. Mean duration of PN/I.V. dependency prior to enrollment was 6.25 years (range 1-25.8 years). The most common reasons for intestinal resection leading to SBS were vascular disease (34.1%, 29/85), Crohn's Disease (21.2%, 18/85), and "other" (21.2%, 18/85). Stoma was present in 44.7% (38/85) of subjects, and the most common type was jejunostomy/ileostomy (81.6%, 31/38). The mean length of remaining small intestine was 77.3±64.4 cm (range: 5 to 343 cm). The colon was not in continuity in 43.5% (37/85) subjects. At baseline, the mean (± SD) prescribed days per week for PN/I.V. infusion was 5.73 (±1.59) days.
- The percentages of treatment group responders were compared in the intent-to-treat population of this study which was defined as all randomized patients. 63% (27/43) of GATTEX-treated subjects versus 30% (13/43) of placebo-treated subjects were considered responders (p=0.002).
- At Week 24, the mean reduction in weekly PN/I.V. volume was 4.4 Liters for GATTEX-treated subjects (from pre-treatment baseline of 12.9 Liters) versus 2.3 Liters for placebo-treated subjects (from pre-treatment baseline of 13.2 Liters/week) (p<0.001).
- Twenty-one subjects on GATTEX (53.8%) versus 9 on placebo (23.1%) achieved at least a one-day reduction in PN/I.V. support.
- The mean changes from Baseline in PN/I.V. volume by visit are shown in Figure 2.
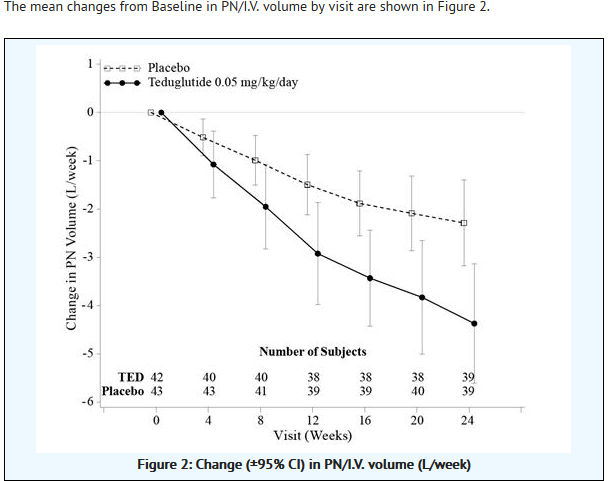
This image is provided by the National Library of Medicine.

- Study 2. Study 2 is an ongoing two-year open-label extension of Study 1 in which 88 subjects receive GATTEX 0.05 mg/kg/day. Ninety-seven percent (76/78) of subjects from Study 1 elected to enroll in Study 2. An additional 12 subjects entered Study 2, who had been optimized and stabilized but not randomized in Study 1 because of closed enrollment. Of responders in Study 1 who entered Study 2, 100% (25/25) sustained their response to GATTEX after one year of continuous treatment. A 20% or greater reduction of parenteral support was achieved in 72% (31/43) of subjects after an additional 28 weeks of continuous GATTEX treatment. The mean reduction of weekly PN/I.V. volume was 5.2 L/week after one year of continuous GATTEX treatment. Six subjects in Study 2 were weaned off their PN/I.V. support while on GATTEX. Subjects were maintained on GATTEX even if no longer requiring PN/I.V. support. These 6 subjects had required PN/I.V. support for 3 to 18 years, and prior to GATTEX had required between 4 L/week and 13 L/week of PN/I.V. support.
Study 3 (Placebo-controlled) and Study 4 (Blinded uncontrolled extension of Study 3)
- Study 3. Study 3 was a randomized, double-blind, placebo-controlled, three parallel-group, multinational study in adults with Short Bowel Syndrome who were dependent on parenteral nutrition/intravenous (PN/I.V.) support for at least 12 months and required PN at least 3 times per week. After a period of optimization and stabilization similar to Study 1, subjects were randomized to receive 24 weeks of one of the following treatment regimens: GATTEX 0.05 mg/kg/day (n=35), GATTEX 0.10 mg/kg/day dose (n=33), or placebo (n=16). The treatment groups were compared using the intent-to-treat population of this study which was defined as all randomized patients who were administered at least one dose of study drug. This population contained one less patient in the 0.10 mg/kg/day dose group hence n=32 in this group for all analyses. The primary efficacy endpoint was a graded categorical score that did not achieve statistical significance for the high dose. Further evaluation of PN/I.V. volume reduction using the endpoint of response (defined as at least 20% reduction in PN/I.V. fluid from Baseline to Weeks 20 and 24) showed that 46% of subjects on GATTEX 0.05 mg/kg/day responded versus 6% on placebo. Subjects on GATTEX at both dose levels experienced a 2.5 L/week reduction in parenteral support requirements versus 0.9 L/week for placebo at 24 weeks. Two subjects in the GATTEX 0.05 mg/kg/day dose group were weaned off parenteral support by Week 24.
- Study 4. Study 4 was a blinded, uncontrolled extension of Study 3, in which 65 subjects from Study 3 received GATTEX for up to an additional 28 weeks of treatment. Of responders in Study 3 who entered Study 4, 75% sustained response on GATTEX after one year of treatment. In the GATTEX 0.05 mg/kg/day dose group, a 20% or greater reduction of parenteral support was achieved in 68% (17/25) of subjects. The mean reduction of weekly PN/I.V. volume was 4.9 L/week (52% reduction from baseline) after one year of continuous GATTEX treatment. The subjects who had been completely weaned off PN/I.V. support in Study 3 remained off parenteral support through Study 4. During Study 4, an additional subject from Study 3 was weaned off parenteral support.
How Supplied
- GATTEX® (teduglutide [rDNA origin]) for injection is supplied in a sterile, single-use glass vial containing 5 mg of teduglutide as a white, lyophilized powder to be reconstituted with 0.5 mL Sterile Water for Injection. The product to be dispensed is either a one-vial kit or a 30-vial kit. The one-vial kit is pre-assembled and ready to be used. The 30-vial kit is to be assembled by a pharmacist with the following two cartons:
- Carton of Drug Vials (NDC 68875-0101-2):
- Thirty single-use vials of drug (NDC 68875-0101-1)
- Carton of Ancillary Supplies:
- Thirty disposable prefilled syringes containing diluent (0.5 mL Sterile Water for Injection USP) for reconstitution
- Thirty separate needles (22G x 1½ in) to attach to the syringes for reconstitution
- Thirty sterile disposable 1-mL syringes with needle (26G x 5/8 in)
- Sixty alcohol swabs
- The pharmacist in a dispensing pharmacy will assemble a 30-vial kit by transferring the trays containing 30 vials from a Carton of Drug Vials into a Carton of Ancillary Supplies. The final patient kits should contain the items listed as follows:
- 30-vial kit (NDC 68875-0102-1):
- Thirty single-use vials of drug (NDC 68875-0101-1)
- Thirty disposable prefilled syringes containing 0.5 mL Sterile Water for Injection USP for reconstitution, with 30 separate needles (22G x 1½ in) to attach to the syringes
- Thirty sterile disposable 1-mL syringes with needle (26G x 5/8 in) for dosing
- Sixty alcohol swabs
- One-vial kit (NDC 68875-0103-1):
- One single-use vial of drug (NDC 68875-0101-1)
- One disposable prefilled syringe containing 0.5 mL Sterile Water for Injection USP for reconstitution, with a separate needle (22G x 1½ in) to attach to the syringe
- One sterile disposable 1-mL syringe with needle (26G x 5/8 in) for dosing
- Four alcohol swabs
- Reconstitution with 0.5 mL of preservative-free Sterile Water for Injection, provided in a prefilled syringe, is required prior to subcutaneous administration of the drug. Reconstituted GATTEX is a sterile, clear, colorless to light straw-colored 10 mg/mL solution, which should be free from particulates. Upon reconstitution with the 0.5 mL Sterile Water for Injection provided in the prefilled syringe, a maximum of 0.38 mL of the reconstituted solution which contains 3.8 mg of teduglutide can be withdrawn from the vial for dosing.
- Storage and Handling
- Prior to Dispensing: Store refrigerated at 2°C to 8°C (36°F to 46°F) for Cartons of Drug Vials and the One-vial kits. Do not freeze. Do not use beyond the expiration date on the label. Store at room temperature up to 25°C (77°F) for the Cartons of Ancillary Supplies.
- Instruction for the Pharmacist:
- Prior to Dispensing: Store at 2°C to 8°C (36°F to 46°F) for Cartons of Drug Vials and the One-vial kits. Do not freeze.
- Dispensing Instructions: Dispense with a 90-day "use by" dating and specify "Store at room temperature up to 25°C (77°F). Do not freeze." Dispense Medication Guide to each patient.
- Reconstituted GATTEX is a sterile, clear, colorless to light straw-colored solution, which should be free from particulates. The drug should be completely dissolved before the solution is withdrawn from the vial. Do not shake or freeze the reconstituted solution. If the product remains undissolved after the second attempt, do not use. GATTEX does not contain any preservatives and is for single-use only. Any unused portion should be discarded. The product should be used within 3 hrs after reconstitution.
Storage
There is limited information regarding Teduglutide Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Teduglutide |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Teduglutide |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- General Counseling Information
- Prior to treatment, patients should fully understand the risks and benefits of GATTEX. Ensure that all patients receive the Medication Guide prior to initiating GATTEX therapy.
- Acceleration of Neoplastic Growth
- Advise patients with active gastrointestinal malignancy (GI tract, hepatobiliary, pancreatic), that GATTEX therapy should be discontinued. In patients with active non-gastrointestinal malignancy, the clinical decision to continue GATTEX should be discussed with patients and be made based on risk-benefit considerations.
- Colorectal polyps.
- Advise patients that colonoscopy of the entire colon with removal of polyps should be done within 6 months prior to starting treatment with GATTEX. A follow-up colonoscopy (or alternate imaging) is recommended at the end of 1 year of GATTEX. Subsequent colonoscopies should be done every 5 years or more often as needed. If a polyp is found, adherence to current polyp follow-up guidelines is recommended. In case of diagnosis of colorectal cancer, GATTEX therapy should be discontinued.
- Small Bowel Neoplasia.
- Advise patients that they should be monitored clinically for small bowel neoplasia. If a benign neoplasm is found, it should be removed. In case of small bowel cancer, GATTEX therapy should be discontinued.
- Intestinal Obstruction
- Advise patients to tell their physician if they experience any signs or symptoms suggestive of intestinal obstruction. If obstruction is present, the physician may temporarily discontinue GATTEX.
- Gallbladder and Bile Duct Disease
- Advise patients that laboratory assessments should be done before and then every 6 months while on GATTEX to monitor gallbladder and biliary function. If clinically significant change occurs, further evaluation (i.e., imaging studies or other) may be necessary. Advise patients to report to their physician all signs and symptoms suggestive of cholecystitis, cholangitis, or cholelithiasis while on GATTEX.
- Pancreatic Disease
- Advise patients that laboratory assessments should be done before and then every 6 months while on GATTEX. If clinically significant change occurs, further evaluation (i.e., imaging studies or other) may be necessary. Advise patients to report to their physician all signs and symptoms suggestive of pancreatic disease while on GATTEX.
- Cardiovascular Disease
- Advise patients with cardiovascular disease to report to their physician any signs of fluid overload or cardiac decompensation while on GATTEX.
- Risks Resulting from Increased Absorption of Concomitant Oral Medication
- Instruct patients to report to all of their physicians any concomitant oral medications that they are taking in order to assess any potential for increased absorption during GATTEX treatment of those oral medications requiring titration or with a narrow therapeutic index.
- Instructions
- Inform patients that GATTEX should not be administered intravenously or intramuscularly. The drug should be used for subcutaneous injection within 3 hours after reconstitution. Advise patients that subcutaneous administration has been associated with injection site reactions, but if they experience a severe reaction including severe rash, they should contact their physician.
- Advise patients that while they may experience abdominal pain and swelling of their stoma especially when starting therapy with GATTEX, if they experience symptoms of intestinal obstruction, they should contact their physician.
- Instruct patients to read the Medication Guide as they are starting GATTEX therapy and to re-read it each time their prescription is renewed.

This image is provided by the National Library of Medicine.

Precautions with Alcohol
- Alcohol-Teduglutide interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- GATTEX®[1]
Look-Alike Drug Names
There is limited information regarding Teduglutide Look-Alike Drug Names in the drug label.
Drug Shortage Status
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Page Name=Teduglutide
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Teduglutide |Label Name=Teduglutide03.png
}}
{{#subobject:
|Label Page=Teduglutide |Label Name=Teduglutide04.png
}}
{{#subobject:
|Label Page=Teduglutide |Label Name=Teduglutide05.png
}}
{{#subobject:
|Label Page=Teduglutide |Label Name=Teduglutide06.png
}}