Regorafenib
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Disclaimer
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
|
WARNING: HEPATOTOXICITY
See full prescribing information for complete Boxed Warning.
* Severe and sometimes fatal hepatotoxicity has been observed in clinical trials.
|
Overview
Regorafenib is an antineoplastic that is FDA approved for the treatment of colorectal cancer, gastrointestinal stromal tumors. There is a Black Box Warning for this drug as shown here. Common adverse reactions include hypertension, mucositis, dysphonia, infection, pain.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Colorectal Cancer
- Regorafenib® is indicated for the treatment of patients with metastatic colorectal cancer (CRC) who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and, if KRAS wild type, an anti-EGFR therapy.
Gastrointestinal Stromal Tumors
- Regorafenib is indicated for the treatment of patients with locally advanced, unresectable or metastatic gastrointestinal stromal tumor (GIST) who have been previously treated with imatinib mesylate and sunitinib malate.
Recommended Dose
- The recommended dose is 160 mg regorafenib (four 40 mg tablets) taken orally once daily for the first 21 days of each 28-day cycle. Continue treatment until disease progression or unacceptable toxicity.
- Take Regorafenib at the same time each day. Swallow tablet whole with a low-fat breakfast that contains less than 30% fat. Examples of a low-fat breakfast include 2 slices of white toast with 1 tablespoon of low-fat margarine and 1 tablespoon of jelly, and 8 ounces of skim milk (319 calories and 8.2 g fat); or 1 cup of cereal, 8 ounces of skim milk, 1 slice of toast with jam, apple juice, and 1 cup of coffee or tea (520 calories and 2 g fat). Do not take two doses of Regorafenib on the same day to make up for a missed dose from the previous day.
Dose Modifications
- Interrupt Regorafenib for the following
- NCI CTCAE Grade 2 hand-foot skin reaction (HFSR) [palmar-plantar erythrodysesthesia (PPE)] that is recurrent or does not improve within 7 days despite dose reduction; interrupt therapy for a minimum of 7 days for Grade 3 HFSR
Symptomatic Grade 2 hypertension
- Any NCI CTCAE Grade 3 or 4 adverse reaction
- Reduce the dose of Regorafenib to 120 mg:
- For the first occurrence of Grade 2 HFSR of any duration
- After recovery of any Grade 3 or 4 adverse reaction
- For Grade 3 aspartate aminotransferase (AST)/alanine aminotransferase (ALT) elevation; only resume if the potential benefit outweighs the risk of hepatotoxicity
- Reduce the dose of Regorafenib to 80 mg
- For re-occurrence of Grade 2 HFSR at the 120 mg dose
- After recovery of any Grade 3 or 4 adverse reaction at the 120 mg dose (except hepatotoxicity)
- Discontinue Regorafenib permanently for the following
- Failure to tolerate 80 mg dose
- Any occurrence of AST or ALT more than 20 times the upper limit of normal (ULN)
- Any occurrence of AST or ALT more than 3 times ULN with concurrent bilirubin more than 2 times ULN
- Re-occurrence of AST or ALT more than 5 times ULN despite dose reduction to 120 mg
- For any Grade 4 adverse reaction; only resume if the potential benefit outweighs the risks
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Regorafenib in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Regorafenib in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Regorafenib in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Regorafenib in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Regorafenib in pediatric patients.
Contraindications
There is limited information regarding Regorafenib Contraindications in the drug label.
Warnings
|
WARNING: HEPATOTOXICITY
See full prescribing information for complete Boxed Warning.
* Severe and sometimes fatal hepatotoxicity has been observed in clinical trials.
|
Hepatotoxicity
- Severe drug induced liver injury with fatal outcome occurred in 0.3% of 1200 Regorafenib-treated patients across all clinical trials. Liver biopsy results, when available, showed hepatocyte necrosis with lymphocyte infiltration. In Study 1, fatal hepatic failure occurred in 1.6% of patients in the regorafenib arm and in 0.4% of patients in the placebo arm; all the patients with hepatic failure had metastatic disease in the liver. In Study 2, fatal hepatic failure occurred in 0.8% of patients in the regorafenib arm.
- Obtain liver function tests (ALT, AST and bilirubin) before initiation of Regorafenib and monitor at least every two weeks during the first 2 months of treatment. Thereafter, monitor monthly or more frequently as clinically indicated. Monitor liver function tests weekly in patients experiencing elevated liver function tests until improvement to less than 3 times the ULN or baseline.
- Temporarily hold and then reduce or permanently discontinue Regorafenib depending on the severity and persistence of hepatotoxicity as manifested by elevated liver function tests or hepatocellular necrosis.
Hemorrhage
- Regorafenib caused an increased incidence of hemorrhage. The overall incidence (Grades 1-5) was 21% and 11% in Regorafenib-treated patients compared to 8% and 3% in placebo-treated patients in Studies 1 and 2. Fatal hemorrhage occurred in 4 of 632 (0.6%) of Regorafenib-treated patients in Studies 1 and 2 and involved the respiratory, gastrointestinal, or genitourinary tracts.
- Permanently discontinue Regorafenib in patients with severe or life-threatening hemorrhage. Monitor INR levels more frequently in patients receiving warfarin.
Dermatological Toxicity
- Regorafenib caused increased incidences of adverse reactions involving the skin and subcutaneous tissues (72% versus 24% in Study 1 and 78% versus 24% in Study 2), including hand-foot skin reaction (HFSR) also known as palmar-plantar erythrodysesthesia (PPE), and severe rash requiring dose modification.
- The overall incidence of HFSR was higher in Regorafenib-treated patients, (45% versus 7% in Study 1 and 67% versus 12% in Study 2), than in the placebo-treated patients. Most cases of HFSR in Regorafenib-treated patients appeared during the first cycle of treatment (69% and 71% of patients who developed HFSR in Study 1 and Study 2, respectively). The incidence of Grade 3 HFSR (17% versus 0% in Study 1 and 22% versus 0% in Study 2), Grade 3 rash (6% versus <1% in Study 1 and 7% versus 0% in Study 2), serious adverse reactions of erythema multiforme (0.2% vs. 0% in Study 1) and Stevens Johnson Syndrome (0.2% vs. 0% in Study 1) was higher in Regorafenib-treated patients.
- Toxic epidermal necrolysis occurred in 0.17% of 1200 Regorafenib-treated patients across all clinical trials.
- Withhold Regorafenib, reduce the dose, or permanently discontinue Regorafenib depending on the severity and persistence of dermatologic toxicity. Institute supportive measures for symptomatic relief.
Hypertension
- Regorafenib caused an increased incidence of hypertension (30% versus 8% in Study 1 and 59% versus 27% in Study 2). Hypertensive crisis occurred in 0.25% of 1200 Regorafenib-treated patients across all clinical trials. The onset of hypertension occurred during the first cycle of treatment in most patients who developed hypertension (72% in Study 1 and Study 2).
- Do not initiate Regorafenib unless blood pressure is adequately controlled. Monitor blood pressure weekly for the first 6 weeks of treatment and then every cycle, or more frequently, as clinically indicated. Temporarily or permanently withhold Regorafenib for severe or uncontrolled hypertension.
Cardiac Ischemia and Infarction
- Regorafenib increased the incidence of myocardial ischemia and infarction in Study 1 (1.2% versus 0.4%). Withhold Regorafenib in patients who develop new or acute onset cardiac ischemia or infarction. Resume Regorafenib only after resolution of acute cardiac ischemic events, if the potential benefits outweigh the risks of further cardiac ischemia.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by characteristic finding on MRI, occurred in one of 1200 Regorafenib-treated patients across all clinical trials. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Discontinue Regorafenib in patients who develop RPLS.
Gastrointestinal Perforation or Fistula
- Gastrointestinal perforation or fistula occurred in 0.6% of 1200 patients treated with Regorafenib across all clinical trials; this included four fatal events. In Study 2, 2.1% (4/188) of Regorafenib-treated patients who were treated during the blinded or open-label portion of the study developed gastrointestinal fistula or perforation; of these, two cases of gastrointestinal perforation were fatal. Permanently discontinue Regorafenib in patients who develop gastrointestinal perforation or fistula.
Wound Healing Complications
- No formal studies of the effect of regorafenib on wound healing have been conducted. Since vascular endothelial growth factor receptor (VEGFR) inhibitors such as regorafenib can impair wound healing, treatment with regorafenib should be stopped at least 2 weeks prior to scheduled surgery. The decision to resume regorafenib after surgery should be based on clinical judgment of adequate wound healing. Regorafenib should be discontinued in patients with wound dehiscence.
Embryo-Fetal Toxicity
- Regorafenib can cause fetal harm when administered to a pregnant woman. Regorafenib was embryolethal and teratogenic in rats and rabbits at exposures lower than human exposures at the recommended dose, with increased incidences of cardiovascular, genitourinary, and skeletal malformations. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus
Adverse Reactions
Clinical Trials Experience
- The following serious adverse reactions are discussed elsewhere in the labeling:
- Hepatotoxicity
- Hemorrhage
- Dermatological Toxicity
- Hypertension
- Cardiac Ischemia and Infarction
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
- Gastrointestinal Perforation or Fistula
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rate observed in practice.
- The most frequently observed adverse drug reactions (≥20%) in patients receiving Regorafenib are asthenia/fatigue, HFSR, diarrhea, decreased appetite/food intake, hypertension, mucositis, dysphonia, infection, pain (not otherwise specified), decreased weight, gastrointestinal and abdominal pain, rash, fever, and nausea.
- The most serious adverse drug reactions in patients receiving Regorafenib are hepatotoxicity, hemorrhage, and gastrointestinal perforation.
Clinical Trials Experience
Colorectal Cancer
- The safety data described below, except where noted, are derived from a randomized (2:1), double-blind, placebo-controlled trial (Study 1) in which 500 patients (median age 61 years; 61% men) with previously-treated metastatic colorectal cancer received Regorafenib as a single agent at the dose of 160 mg daily for the first 3 weeks of each 4 week treatment cycle and 253 patients (median age 61 years; 60% men) received placebo. The median duration of therapy was 7.3 (range 0.3, 47.0) weeks for patients receiving Regorafenib. Due to adverse reactions, 61% of the patients receiving Regorafenib required a dose interruption and 38% of the patients had their dose reduced. Drug-related adverse reactions that resulted in treatment discontinuation were reported in 8.2% of Regorafenib-treated patients compared to 1.2% of patients who received placebo. Hand-foot skin reaction (HFSR) and rash were the most common reasons for permanent discontinuation of Regorafenib.
- TABLE 1 compares the incidence of adverse reactions (≥10%) in patients receiving Regorafenib and reported more commonly than in patients receiving placebo (Study 1).

This image is provided by the National Library of Medicine.

Postmarketing Experience
There is limited information regarding Postmarketing Experience of Regorafenib in the drug label.
Drug Interactions
Effect of Strong CYP3A4 Inducers on Regorafenib
- Co-administration of a strong CYP3A4 inducer (rifampin) with a single 160 mg dose of Regorafenib decreased the mean exposure of regorafenib, increased the mean exposure of the active metabolite M-5, and resulted in no change in the mean exposure of the active metabolite M-2. Avoid concomitant use of Regorafenib with strong CYP3A4 inducers (e.g. rifampin, phenytoin, carbamazepine, phenobarbital, and St. John’s Wort).
Effect of Strong CYP3A4 Inhibitors on Regorafenib
- Co-administration of a strong CYP3A4 inhibitor (ketoconazole) with a single 160 mg dose of Regorafenib increased the mean exposure of regorafenib and decreased the mean exposure of the active metabolites M-2 and M-5. Avoid concomitant use of Regorafenib with strong inhibitors of CYP3A4 activity (e.g. clarithromycin, grapefruit juice, itraconazole, ketoconazole, nefazodone, posaconazole, telithromycin, and voriconazole).
Use in Specific Populations
Pregnancy
Risk Summary
- Based on its mechanism of action, Regorafenib can cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies with Regorafenib in pregnant women. Regorafenib was embryolethal and teratogenic in rats and rabbits at exposures lower than human exposures at the recommended dose, with increased incidences of cardiovascular, genitourinary, and skeletal malformations. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus.
Animal Data
- In embryo-fetal development studies, a total loss of pregnancy (100% resorption of litter) was observed in rats at doses as low as 1 mg/kg (approximately 6% of the recommended human dose, based on body surface area) and in rabbits at doses as low as 1.6 mg/kg (approximately 25% of the human exposure at the clinically recommended dose measured by AUC).
- In a single dose distribution study in pregnant rats, there was increased penetration of regorafenib across the blood-brain barrier in fetuses compared to dams. In a repeat dose study with daily administration of regorafenib to pregnant rats during organogenesis, findings included delayed ossification in fetuses at doses > 0.8 mg/kg (approximately 5% of the recommended human dose based on body surface area) with dose-dependent increases in skeletal malformations including cleft palate and enlarged fontanelle at doses ≥ 1 mg/kg (approximately 10% of the clinical exposure based on AUC). At doses ≥ 1.6 mg/kg (approximately 11% of the recommended human dose based on body surface area), there were dose-dependent increases in the incidence of cardiovascular malformations, external abnormalities, diaphragmatic hernia, and dilation of the renal pelvis.
- In pregnant rabbits administered regorafenib daily during organogenesis, there were findings of ventricular septal defects evident at the lowest tested dose of 0.4 mg/kg (approximately 7% of the AUC in patients at the recommended dose). At doses of ≥ 0.8 mg/kg (approximately 15% of the human exposure at the recommended human dose based on AUC), administration of regorafenib resulted in dose-dependent increases in the incidence of additional cardiovascular malformations and skeletal anomalies as well as significant adverse effects on the urinary system including missing kidney/ureter; small, deformed and malpositioned kidney; and hydronephrosis. The proportion of viable fetuses that were male decreased with increasing dose in two rabbit embryo-fetal toxicity studies.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Regorafenib in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Regorafenib during labor and delivery.
Nursing Mothers
- It is unknown whether regorafenib or its metabolites are excreted in human milk. In rats, regorafenib and its metabolites are excreted in milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Regorafenib, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
- The safety and efficacy of Regorafenib in pediatric patients less than 18 years of age have not been established.
- In 28-day repeat dose studies in rats there were dose-dependent findings of dentin alteration and angiectasis. These findings were observed at regorafenib doses as low as 4 mg/kg (approximately 25% of the AUC in humans at the recommended dose). In 13-week repeat dose studies in dogs there were similar findings of dentin alteration at doses as low as 20 mg/kg (approximately 43% of the AUC in humans at the recommended dose).
- Administration of regorafenib in these animals also led to persistent growth and thickening of the femoral epiphyseal growth plate.
Geriatic Use
- Of the 632 Regorafenib-treated patients enrolled in Studies 1 and 2, 37% were 65 years of age or and over, while 8% were 75 and over. No overall differences in safety or efficacy were observed between these patients and younger patients.
Gender
There is no FDA guidance on the use of Regorafenib with respect to specific gender populations.
Race
There is no FDA guidance on the use of Regorafenib with respect to specific racial populations.
Renal Impairment
- No clinically relevant differences in the mean exposure of regorafenib and the active metabolites M-2 and M-5 were observed in patients with mild renal impairment (CLcr 60-89 mL/min) compared to patients with normal renal function following regorafenib 160 mg daily for 21 days. No dose adjustment is recommended for patients with mild renal impairment.
- Limited pharmacokinetic data are available from patients with moderate renal impairment (CLcr 30-59 mL/min). Regorafenib has not been studied in patients with severe renal impairment or end-stage renal disease.
Hepatic Impairment
- No clinically important differences in the mean exposure of regorafenib or the active metabolites M-2 and M-5 were observed in patients with hepatocellular carcinoma and mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment compared to patients with normal hepatic functions.
- No dose adjustment is recommended in patients with mild or moderate hepatic impairment. Closely monitor patients with hepatic impairment for adverse reactions.
- Regorafenib is not recommended for use in patients with severe hepatic impairment (Child-Pugh Class C), as it has not been studied in this population.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Regorafenib in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Regorafenib in patients who are immunocompromised.
Administration and Monitoring
Administration
Monitoring
There is limited information regarding Monitoring of Regorafenib in the drug label.
- Description
IV Compatibility
There is limited information regarding IV Compatibility of Regorafenib in the drug label.
Overdosage
- The highest dose of Regorafenib studied clinically is 220 mg per day. In the event of suspected overdose, interrupt Regorafenib, institute supportive care, and observe until clinical stabilization.
Pharmacology

Mechanism of Action
- Regorafenib is a small molecule inhibitor of multiple membrane-bound and intracellular kinases involved in normal cellular functions and in pathologic processes such as oncogenesis, tumor angiogenesis, and maintenance of the tumor microenvironment. In in vitro biochemical or cellular assays, regorafenib or its major human active metabolites M-2 and M-5 inhibited the activity of RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, BRAF, BRAFV600E , SAPK2, PTK5, and Abl at concentrations of regorafenib that have been achieved clinically.
- In in vivo models, regorafenib demonstrated anti-angiogenic activity in a rat tumor model, and inhibition of tumor growth as well as anti-metastatic activity in several mouse xenograft models including some for human colorectal carcinoma.
Structure
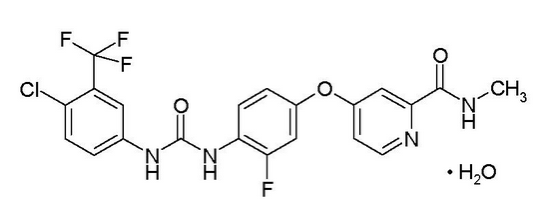
This image is provided by the National Library of Medicine.
Pharmacodynamics
There is limited information regarding Pharmacodynamics of Regorafenib in the drug label.
Pharmacokinetics
Absorption
- Following a single 160 mg dose of Regorafenib in patients with advanced solid tumors, regorafenib reaches a geometric mean peak plasma level (Cmax) of 2.5 µg/mL at a median time of 4 hours and a geometric mean area under the plasma concentration vs. time curve (AUC) of 70.4 µg*h/mL. The AUC of regorafenib at steady-state increases less than dose proportionally at doses greater than 60 mg. At steady-state, regorafenib reaches a geometric mean Cmax of 3.9 µg/mL and a geometric mean AUC of 58.3 µg*h/mL. The coefficient of variation of AUC and Cmax is between 35% and 44%.
- The mean relative bioavailability of tablets compared to an oral solution is 69% to 83%.
- In a food-effect study, 24 healthy men received a single 160 mg dose of Regorafenib on three separate occasions: under a fasted state, with a high-fat meal and with a low-fat meal. A high-fat meal (945 calories and 54.6 g fat) increased the mean AUC of regorafenib by 48% and decreased the mean AUC of the M-2 and M-5 metabolites by 20% and 51%, respectively, as compared to the fasted state. A low-fat meal (319 calories and 8.2 g fat) increased the mean AUC of regorafenib, M-2 and M-5 by 36%, 40% and 23%, respectively as compared to fasted conditions. Regorafenib was administered with a low-fat meal in Studies 1 and 2.
Distribution
- Regorafenib undergoes enterohepatic circulation with multiple plasma concentration peaks observed across the 24-hour dosing interval. Regorafenib is highly bound (99.5%) to human plasma proteins.
Metabolism
- Regorafenib is metabolized by CYP3A4 and UGT1A9. The main circulating metabolites of regorafenib measured at steady-state in human plasma are M-2 (N-oxide) and M-5 (N-oxide and N-desmethyl), both of them having similar in vitro pharmacological activity and steady-state concentrations as regorafenib. M-2 and M-5 are highly protein bound (99.8% and 99.95%, respectively).
Elimination
- Following a single 160 mg oral dose of Regorafenib, the geometric mean (range) elimination half-lives for regorafenib and the M-2 metabolite in plasma are 28 hours (14 to 58 hours) and 25 hours (14 to 32 hours), respectively. M-5 has a longer mean (range) elimination half-life of 51 hours (32 to 70 hours).
- Approximately 71% of a radiolabeled dose was excreted in feces (47% as parent compound, 24% as metabolites) and 19% of the dose was excreted in urine (17% as glucuronides) within 12 days after administration of a radiolabeled oral solution at a dose of 120 mg.
Age, Gender, and Weight
- Based on the population pharmacokinetic analysis, there is no clinically relevant effect of age, gender or weight on the pharmacokinetics of regorafenib.
Hepatic Impairment
- The pharmacokinetics of regorafenib, M-2, and M-5 was evaluated in 14 patients with hepatocellular carcinoma (HCC) and mild hepatic impairment (Child-Pugh A); 4 patients with HCC and moderate hepatic impairment (Child-Pugh B); and 10 patients with solid tumors and normal hepatic function after the administration of a single 100 mg dose of Regorafenib. No clinically important differences in the mean exposure of regorafenib, M-2, or M-5 were observed in patients with mild or moderate hepatic impairment compared to the patients with normal hepatic function. The pharmacokinetics of regorafenib has not been studied in patients with severe hepatic impairment (Child-Pugh C).
Renal Impairment
- The pharmacokinetics of regorafenib, M-2, and M-5 was evaluated in 10 patients with mild renal impairment (CLcr 60-89 mL/min) and 18 patients with normal renal function following the administration of Regorafenib at a dose of 160 mg daily for 21 days. No differences in the mean steady-state exposure of regorafenib, M-2, or M-5 were observed in patients with mild renal impairment compared to patients with normal renal function. Limited pharmacokinetic data are available from patients with moderate renal impairment (CLcr 30-59 mL/min). The pharmacokinetics of regorafenib has not been studied in patients with severe renal impairment or end-stage renal disease.
Drug-Drug Interactions
- Effect of Regorafenib on Cytochrome P450 Substrates
- In vitro studies suggested that regorafenib is an inhibitor of CYP2C8, CYP2C9, CYP2B6, CYP3A4 and CYP2C19; M-2 metabolite is an inhibitor of CYP2C9, CYP2C8, CYP3A4 and CYP2D6, and M-5 metabolite is an inhibitor of CYP2C8. In vitro studies suggested that regorafenib is not an inducer of CYP1A2, CYP2B6, CYP2C19, and CYP3A4 enzyme activity.
- Patients with advanced solid tumors received single oral doses of CYP substrates, 2 mg of midazolam (CYP3A4), 40 mg of omeprazole (CYP2C19) and 10 mg of warfarin (CYP2C9) or 4 mg of rosiglitazone (CYP2C8) one week before and two weeks after Regorafenib at a dose of 160 mg once daily. No clinically relevant change was observed in the mean AUC of rosiglitazone (N=12) or the mean omeprazole (N=11) plasma concentrations measured 6 hours after dosing or the mean AUC of midazolam (N=15). The mean AUC of warfarin (N=8) increased by 25%.
- Effect of CYP3A4 Strong Inducers on Regorafenib: Twenty-two healthy men received a single 160 mg dose of Regorafenib alone and then 7 days after starting rifampin. Rifampin, a strong CYP3A4 inducer, was administered at a dose of 600 mg daily for 9 days. The mean AUC of regorafenib decreased by 50% and mean AUC of M-5 increased by 264%. No change in the mean AUC of M-2 was observed.
- Effect of CYP3A4 Strong Inhibitors on Regorafenib: Eighteen healthy men received a single 160 mg dose of Regorafenib alone and then 5 days after starting ketoconazole. Ketoconazole, a strong CYP3A4 inhibitor, was administered at a dose of 400 mg daily for 18 days. The mean AUC of regorafenib increased by 33% and the mean AUC of M-2 and M-5 both decreased by 93%.
- Effect of Regorafenib on UGT1A1 Substrates: In vitro studies showed that regorafenib, M-2, and M-5 competitively inhibit UGT1A9 and UGT1A1 at therapeutically relevant concentrations. Eleven patients received irinotecan-containing combination chemotherapy with Regorafenib at a dose of 160 mg. The mean AUC of irinotecan increased 28% and the mean AUC of SN-38 increased by 44% when irinotecan was administered 5 days after the last of 7 daily doses of Regorafenib.
- In vitro screening of transporters: In vitro data suggested that regorafenib is an inhibitor of ABCG2 (Breast Cancer Resistance Protein) and ABCB1 (P-glycoprotein).
Cardiac Electrophysiology
- The effect of multiple doses of Regorafenib (160 mg once daily for 21 days) on the QTc interval was evaluated in an open label, single arm study in 25 patients with advanced solid tumors. No large changes in the mean QTc interval (i.e., > 20 msec) were detected in the study.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment of Fertility
- Studies examining the carcinogenic potential of regorafenib have not been conducted. Regorafenib itself did not demonstrate genotoxicity in in vitro or in vivo assays; however, a major human active metabolite of regorafenib, (M-2), was positive for clastogenicity, causing chromosome aberration in Chinese hamster V79 cells.
- Dedicated studies to examine the effects of regorafenib on fertility have not been conducted; however, there were histological findings of tubular atrophy and degeneration in the testes, atrophy in the seminal vesicle, and cellular debris and oligospermia in the epididymides in male rats at doses similar to those in human at the clinical recommended dose based on AUC. In female rats, there were increased findings of necrotic corpora lutea in the ovaries at the same exposures. There were similar findings in dogs of both sexes in repeat dose studies at exposures approximately 83% of the human exposure at the recommended human dose based on AUC. These findings suggest that regorafenib may adversely affect fertility in humans.
Animal Toxicology and/or Pharmacology
- In a chronic 26-week repeat dose study in rats there was a dose-dependent increase in the finding of thickening of the atrioventricular valve. At a dose that resulted in an exposure of approximately 12% of the human exposure.
Clinical Studies
Colorectal Cancer
- The clinical efficacy and safety of Regorafenib were evaluated in an international, multi-center, randomized (2:1), double-blind, placebo-controlled trial (Study 1) in 760 patients with previously-treated metastatic colorectal cancer. The major efficacy outcome measure was overall survival (OS); supportive efficacy outcome measures included progression-free survival (PFS) and objective tumor response rate.
- Patients were randomized to receive 160 mg regorafenib orally once daily (N=505) plus Best Supportive Care (BSC) or placebo (N=255) plus BSC for the first 21 days of each 28-day cycle. Regorafenib was administered with a low-fat breakfast that contains less than 30% fat. Treatment continued until disease progression or unacceptable toxicity.
- In the all-randomized population, median age was 61 years, 61% were men, 78% were White, and all patients had baseline ECOG performance status of 0 or 1. The primary site of disease was colon (65%), rectum (29%), or both (6%). History of KRAS evaluation was reported for 729 (96%) patients; 430 (59%) of these patients were reported to have KRAS mutation. The median number of prior lines of therapy for metastatic disease was 3. All patients received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, and with bevacizumab. All but one patient with KRAS mutation-negative tumors received panitumumab or cetuximab.

This image is provided by the National Library of Medicine.

Gastrointestinal Stromal Tumors
- The efficacy and safety of Regorafenib were evaluated in an international, multi-center, randomized (2:1), double-blind, placebo-controlled trial (Study 2) in 199 patients with unresectable, locally advanced or metastatic gastrointestinal stromal tumor (GIST), who had been previously treated with imatinib mesylate and sunitinib malate. Randomization was stratified by line of therapy (third vs. four or more) and geographic region (Asia vs. rest of the world).
- The major efficacy outcome measure of Study 2 was progression-free survival (PFS) based on disease assessment by independent radiological review using modified RECIST 1.1 criteria, in which lymph nodes and bone lesions were not target lesions and progressively growing new tumor nodule within a pre-existing tumor mass was progression. The key secondary outcome measure was overall survival.
- Patients were randomized to receive 160 mg regorafenib orally once daily (N=133) plus best supportive care (BSC) or placebo (N=66) plus BSC for the first 21 days of each 28-day cycle. Treatment continued until disease progression or unacceptable toxicity. In Study 2, the median age of patients was 60 years, 64% were men, 68% were White, and all patients had baseline ECOG performance status of 0 (55%) or 1 (45%). At the time of disease progression as assessed by central review, the study blind was broken and all patients were offered the opportunity to take Regorafenib at the investigator’s discretion. Fifty-six (85%) patients randomized to placebo and 41 (31%) patients randomized to Regorafenib received open-label Regorafenib.
- A statistically significant improvement in PFS was demonstrated among patients treated with Regorafenib compared to placebo (see Table 6 and Figure 2). There was no statistically significant difference in overall survival at the time of the planned interim analysis based on 29% of the total events for the final analysis.
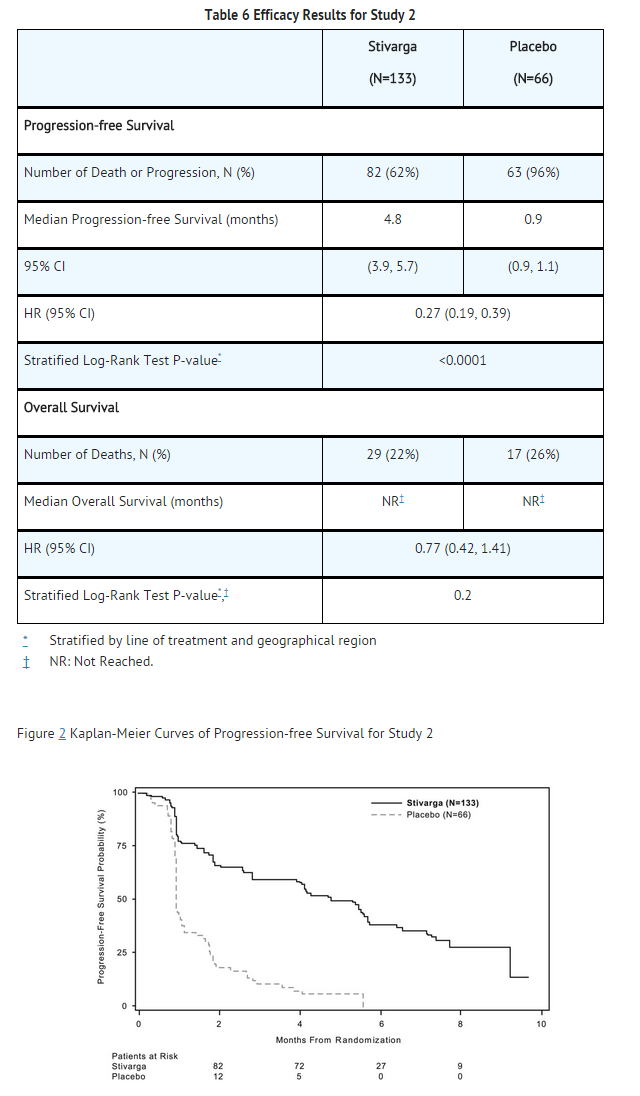
This image is provided by the National Library of Medicine.

How Supplied
- Regorafenib tablets are supplied in packages containing three bottles, with each bottle containing 28 tablets, for a total of 84 tablets per package (NDC 50419-171-03).
Storage
- Store Regorafenib at 25°C (77°F); excursions are permitted from 15 to 30°C (59 to 86°F).
- Store tablets in the original bottle and do not remove the desiccant. Keep the bottle tightly closed after first opening.
- Discard any unused tablets 7 weeks after opening the bottle.Dispose of unused tablets in accordance with local requirements.
Images
Drug Images
{{#ask: Page Name::Regorafenib |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel


{{#ask: Label Page::Regorafenib |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- See FDA-Approved Patient Labeling (Patient Information).
- Inform your patients of the following:
- Regorafenib may cause severe or life-threatening liver damage. Inform patients that they will need to undergo monitoring for liver damage and to immediately report any signs or symptoms of severe liver damage to their health care provider.
- Regorafenib can cause severe bleeding. Advise patients to contact their health care provider for any episode of bleeding.
- Regorafenib can cause hand-foot skin reactions or rash elsewhere. Advise patients to contact their health care provider if they experience skin changes associated with redness, pain, blisters, bleeding, or swelling.
- Regorafenib can cause or exacerbate existing hypertension. Advise patients they will need to undergo blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms.
- Regorafenib increased the risk for myocardial ischemia and infarction. Advise patients to seek immediate emergency help if they experience chest pain, shortness of breath, or feel dizzy or like passing out.
Contact a healthcare provider immediately if they experience severe pains in their abdomen, persistent swelling of the abdomen, high fever, chills, nausea, vomiting, severe diarrhea (frequent or loose bowel movements), or dehydration.
- Regorafenib may complicate wound healing. Advise patients to inform their health care provider if they plan to undergo a surgical procedure or had recent surgery.
- Inform patients that regorafenib can cause fetal harm. Advise women of reproductive potential and men of the need for effective contraception during Regorafenib treatment and for up to 2 months after completion of treatment. Instruct women of reproductive potential to immediately contact her health care provider if pregnancy is suspected or confirmed during or within 2 months of completing treatment with Regorafenib.
- Advise nursing mothers that it is not known whether regorafenib is present in breast milk and discuss whether to discontinue nursing or to discontinue regorafenib.
- Inform patients to take any missed dose on the same day, as soon as they remember, and that they must not take two doses on the same day to make up for a dose missed on the previous day.
- Inform patients to store medicine in the original container. Do not place medication in daily or weekly pill boxes . Any remaining tablets should be discarded 7 weeks after opening the bottle. Tightly close bottle after each opening and keep the desiccant in the bottle.
Precautions with Alcohol
- Alcohol-Regorafenib interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- Regorafenib ®[1]
Look-Alike Drug Names
There is limited information regarding Regorafenib Look-Alike Drug Names in the drug label.
Drug Shortage Status
Price
References
The contents of this FDA label are provided by the National Library of Medicine.