Membranous glomerulonephritis pathophysiology: Difference between revisions
Sunny Kumar (talk | contribs) |
No edit summary |
||
| Line 3: | Line 3: | ||
{{CMG}}; {{AE}} | {{CMG}}; {{AE}} | ||
==Overview== | ==Overview== | ||
It is thought that [disease name] is the result of / is mediated by / is produced by / is caused by either [hypothesis 1], [hypothesis 2], or [hypothesis 3]. | It is thought that [disease name] is the result of / is mediated by / is produced by / is caused by either [hypothesis 1], [hypothesis 2], or [hypothesis 3]. | ||
==Pathophysiology== | |||
Phospholipase A2 receptor — The M-type PLA2R, a transmembrane receptor that is highly expressed in glomerular podocytes, has been identified as a major antigen in human idiopathic MN [22]. In this study, circulating autoantibodies to PLA2R were identified in 26 of 37 (70 percent) patients with idiopathic MN and could be associated with disease activity in patients for whom serial serum samples were available [22]. By contrast, there was no evidence of PLA2R antibodies in serum from eight patients with secondary MN due to lupus or hepatitis B; from 15 patients with proteinuric conditions other than MN (such as diabetic nephropathy or focal segmental glomerulosclerosis); or from 30 healthy control individuals. The circulating anti-PLA2R antibodies were predominantly IgG4, the IgG subclass that is most abundant in the glomerular immune deposits in idiopathic (but not secondary) MN. PLA2R colocalized with IgG4 in immune deposits of kidney tissue obtained by kidney biopsy from patients with MN, and anti-PLA2R antibodies could be eluted from this tissue. This was in contrast to the findings in secondary MN biopsies, in which there was no colocalization of IgG4 and PLA2R and from which no anti-PLA2R antibodies could be eluted. | |||
The majority of anti-PLA2R autoantibodies appear to target a specific region of the PLA2R protein [23-25]. Two independent studies identified three domains near the N-terminus of PLA2R as the dominant epitope for anti-PLA2R [23,24], and one study further identified a small, nine-amino acid sequence that could largely inhibit the antibody-antigen interaction [23]. | |||
Anti-PLA2R antibodies have been identified in 57, 74, 75, 78, 80, and 82 percent of patients with primary MN in six additional cohorts from Europe and China [26-31]. In one study, anti-PLA2R strongly correlated with clinical status [28]; in another study, lower anti-PLA2R titers were associated with a higher rate of spontaneous remission [29], and, in two other studies, a decline in anti-PLA2R predicted the clinical response to immunosuppressive therapy [31,32]. A fifth study found that higher anti-PLA2R titers within two years of diagnosis predicted substantially greater progression of kidney function decline over the subsequent five years of follow-up [30]. However, this may have reflected increased disease activity at the time the serum was collected; lower titers may have identified individuals already undergoing immunological and clinical remission. (See "Treatment of idiopathic membranous nephropathy", section on 'Resistant disease'.) | |||
Staining the kidney biopsy specimen for PLA2R, either by immunofluorescence or immunohistochemistry, provides another assay by which to identify PLA2R-associated primary MN [26,33,34]. As an example, in the study mentioned above that reported a relatively low sensitivity of circulating anti-PLA2R (57 percent), an additional 24 percent of patients who did not have circulating antibodies had the PLA2R antigen detected within immune deposits by immunofluorescence of the biopsy specimen [26]. This may occur as patients enter serological remission with still unresolved proteinuria and persistent immune deposits in glomeruli. Theoretically, it could also occur in the early stages of disease as anti-PLA2R antibodies are "soaked up" in the immune deposits and have not yet reached sufficiently high levels to be detected in the serum by existing immunoassays (figure 1) [35-37]. In general, tissue staining for PLA2R may be more sensitive (69 to 84 percent in various studies) than circulating anti-PLA2R in patients with primary MN [26,33,34,38,39]. Specificity is close to 100 percent; however, PLA2R has been detected in the immune deposits of some patients with secondary MN associated with hepatitis B virus infection, neoplasms, nonsteroidal antiinflammatory drug (NSAID) use, or sarcoidosis, but not systemic lupus nephritis [33,34,38,39]. It is possible that this represents a coincidental association, and we regard such cases as having PLA2R-associated MN. | |||
The detection of circulating anti-PLA2R and/or PLA2R kidney immune deposits in the majority of adult patients with idiopathic MN represents a large step forward in our understanding of the disease as investigators will now be able to test whether the pathogenetic mechanisms learned from experimental models are also involved in human disease. The sensitivity and specificity of anti-PLA2R autoantibodies in individuals with immunologically active idiopathic MN have also enabled the development of a serologic immunoassay for the noninvasive diagnosis of primary MN and monitoring of disease activity. It should be noted that, while the association of these anti-PLA2R autoantibodies with disease suggests a causal role, this has not yet been confirmed experimentally. | |||
Thrombospondin type-1 domain-containing 7A — THSD7A is, like PLA2R, a transmembrane protein expressed on podocytes [40,41]. THSD7A may be the responsible antigen in approximately 10 percent of patients with idiopathic MN who are negative for anti-PLA2R antibodies. The association of THSD7A with MN was examined in a study of 154 patients with anti-PLA2R-negative idiopathic MN, 74 patients with anti-PLA2R-positive idiopathic MN, 76 patients with other glomerular disease, and 44 healthy controls [40]. Autoantibodies specific for THSD7A were identified in sera from 15 of 154 patients with anti-PLA2R-negative idiopathic MN but not in the sera from other individuals. In addition, the IgG that was eluted from the kidney biopsies of 1 of these 15 patients was specific for THSD7A, providing further support that THSD7A was the target antigen in these patients. THSD7A-associated MN has been found at a low frequency in North American and European cohorts, but it may be more prevalent in Japanese patients with primary MN [42]. | |||
THSD7A may also be involved in the pathogenesis of some cases of malignancy-associated MN [43]. (See 'Malignancy' below.) | |||
Neutral endopeptidase — Neutral endopeptidase (NEP), which is expressed on podocytes, is the probable target in a rare antenatal form of MN [44,45]. The transplacental passage of anti-NEP antibodies (from mothers genetically deficient in NEP who were alloimmunized during a prior pregnancy) caused MN with subepithelial immune deposits (anti-NEP and NEP) in the fetus/neonate. Nephrotic syndrome resolved several months after birth, with disappearance of the deposits, upon clearance of the maternal antibodies. Although noncomplement-fixing IgG4 was the predominant IgG subclass of anti-NEP in alloimmunized NEP-deficient mothers, the development of proteinuria in their babies correlated with the additional presence of complement-fixing IgG1 anti-NEP [45]. This finding, together with reports showing that C5b-9 is shed into the urine of patients with recent-onset MN [46,47], provides additional evidence that the observations in Heymann nephritis are relevant to the human disease. It has yet to be determined, however, whether the IgG4 anti-PLA2R antibodies that predominate in idiopathic MN or accompanying IgG1 antibodies are responsible for complement activation. | |||
Intracellular antigens — In addition to the PLA2R, THSD7A, and NEP, antibodies directed against other antigens expressed by podocytes may contribute to the pathogenesis of MN [48-50]. In one study, for example, serum IgG4 reactivity against aldose reductase, superoxide dismutase 2, and alpha-enolase, as well as the PLA2R and neutral endopeptidase, was measured in 186 patients with MN, 36 patients with focal glomerulosclerosis, and 60 patients with immunoglobulin A (IgA) nephropathy [48]. Elevated titers of IgG4 against the PLA2R, alpha-enolase, aldose reductase, and superoxide dismutase 2 were found in 60, 43, 34, and 28 percent of patients with MN, respectively, but not in patients with other glomerular diseases. Approximately one-half of the patients who were negative for antibodies against the PLA2R had an elevated titer for one of the other three antibodies. Although these antigens are predominantly intracellular, it has been proposed that podocyte injury causes the intracellular enzymes to translocate to the cell surface where they are accessible to the circulating antibodies, causing amplification of the immune injury and possibly aggravating the course of the disease. | |||
Cationic bovine serum albumin — Antibodies to a cationic form of bovine serum albumin (BSA) are present in a small number of children with MN [51]. The BSA antigen, which was found within the immune deposits of biopsy specimens from these patients, is thought to be absorbed from the relatively underdeveloped pediatric intestinal tract in a partially digested or undigested form and then serve as a planted antigen within the glomerular capillary wall. Antibodies reactive with bovine, but not human, serum albumin were eluted from the kidney biopsy specimen in one case. | |||
The | |||
Other antigens — Other antigens have been identified within the glomerular immune deposits in patients with secondary MN [52]. These antigens include double-stranded DNA in systemic lupus erythematosus (SLE), thyroglobulin in thyroiditis, hepatitis B antigen, treponemal antigen and Helicobacter pylori in the relevant infections, and carcinoembryonic antigen and prostate specific antigen in malignancy. The pathogenicity of these antigens is unproven. | |||
Role of T cells — T helper cells activate different immune effector mechanisms and appear to play a role in the pathogenesis of glomerulonephritis and may also participate in the genesis of proteinuria in MN. The T helper subset Th1 tends to predominate in proliferative and crescentic forms of glomerulonephritis, whereas Th2 predominates in MN and minimal change disease [53,54]. (See "The adaptive cellular immune response", section on 'Cytokine secretion profiles of activated T cells' and "Mechanisms of immune injury of the glomerulus", section on 'T cells'.) | |||
In support of a pathogenetic role for Th2 in MN is the observation in a model of lupus nephritis that deletion of the ''WSX-1'' gene (encoding a cytokine receptor integral for mounting a Th1 response) causes a shift toward a Th2 response and converts the diffuse proliferative pattern that is typically seen to a membranous pattern [55]. The site of the target antigen and subepithelial location of the immune deposits also favor antibody- and complement-mediated injury to the podocyte, rather than a direct cellular inflammatory lesion (which is typically associated with subendothelial immune deposits and glomerular endothelial injury) [56,57]. | |||
==Genetics== | ==Genetics== | ||
==Associated Conditions== | ==Associated Conditions== | ||
==Gross Pathology== | ==Gross Pathology== | ||
==Microscopic Pathology== | ==Microscopic Pathology== | ||
==References== | ==References== | ||
{{Reflist|2}} | {{Reflist|2}} | ||
Revision as of 11:54, 20 May 2018
|
Membranous glomerulonephritis Microchapters |
|
Differentiating Membranous glomerulonephritis from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Membranous glomerulonephritis pathophysiology On the Web |
|
American Roentgen Ray Society Images of Membranous glomerulonephritis pathophysiology |
|
Directions to Hospitals Treating Membranous glomerulonephritis |
|
Risk calculators and risk factors for Membranous glomerulonephritis pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief:
Overview
It is thought that [disease name] is the result of / is mediated by / is produced by / is caused by either [hypothesis 1], [hypothesis 2], or [hypothesis 3].
Pathophysiology
Phospholipase A2 receptor — The M-type PLA2R, a transmembrane receptor that is highly expressed in glomerular podocytes, has been identified as a major antigen in human idiopathic MN [22]. In this study, circulating autoantibodies to PLA2R were identified in 26 of 37 (70 percent) patients with idiopathic MN and could be associated with disease activity in patients for whom serial serum samples were available [22]. By contrast, there was no evidence of PLA2R antibodies in serum from eight patients with secondary MN due to lupus or hepatitis B; from 15 patients with proteinuric conditions other than MN (such as diabetic nephropathy or focal segmental glomerulosclerosis); or from 30 healthy control individuals. The circulating anti-PLA2R antibodies were predominantly IgG4, the IgG subclass that is most abundant in the glomerular immune deposits in idiopathic (but not secondary) MN. PLA2R colocalized with IgG4 in immune deposits of kidney tissue obtained by kidney biopsy from patients with MN, and anti-PLA2R antibodies could be eluted from this tissue. This was in contrast to the findings in secondary MN biopsies, in which there was no colocalization of IgG4 and PLA2R and from which no anti-PLA2R antibodies could be eluted.
The majority of anti-PLA2R autoantibodies appear to target a specific region of the PLA2R protein [23-25]. Two independent studies identified three domains near the N-terminus of PLA2R as the dominant epitope for anti-PLA2R [23,24], and one study further identified a small, nine-amino acid sequence that could largely inhibit the antibody-antigen interaction [23].
Anti-PLA2R antibodies have been identified in 57, 74, 75, 78, 80, and 82 percent of patients with primary MN in six additional cohorts from Europe and China [26-31]. In one study, anti-PLA2R strongly correlated with clinical status [28]; in another study, lower anti-PLA2R titers were associated with a higher rate of spontaneous remission [29], and, in two other studies, a decline in anti-PLA2R predicted the clinical response to immunosuppressive therapy [31,32]. A fifth study found that higher anti-PLA2R titers within two years of diagnosis predicted substantially greater progression of kidney function decline over the subsequent five years of follow-up [30]. However, this may have reflected increased disease activity at the time the serum was collected; lower titers may have identified individuals already undergoing immunological and clinical remission. (See "Treatment of idiopathic membranous nephropathy", section on 'Resistant disease'.)
Staining the kidney biopsy specimen for PLA2R, either by immunofluorescence or immunohistochemistry, provides another assay by which to identify PLA2R-associated primary MN [26,33,34]. As an example, in the study mentioned above that reported a relatively low sensitivity of circulating anti-PLA2R (57 percent), an additional 24 percent of patients who did not have circulating antibodies had the PLA2R antigen detected within immune deposits by immunofluorescence of the biopsy specimen [26]. This may occur as patients enter serological remission with still unresolved proteinuria and persistent immune deposits in glomeruli. Theoretically, it could also occur in the early stages of disease as anti-PLA2R antibodies are "soaked up" in the immune deposits and have not yet reached sufficiently high levels to be detected in the serum by existing immunoassays (figure 1) [35-37]. In general, tissue staining for PLA2R may be more sensitive (69 to 84 percent in various studies) than circulating anti-PLA2R in patients with primary MN [26,33,34,38,39]. Specificity is close to 100 percent; however, PLA2R has been detected in the immune deposits of some patients with secondary MN associated with hepatitis B virus infection, neoplasms, nonsteroidal antiinflammatory drug (NSAID) use, or sarcoidosis, but not systemic lupus nephritis [33,34,38,39]. It is possible that this represents a coincidental association, and we regard such cases as having PLA2R-associated MN.
The detection of circulating anti-PLA2R and/or PLA2R kidney immune deposits in the majority of adult patients with idiopathic MN represents a large step forward in our understanding of the disease as investigators will now be able to test whether the pathogenetic mechanisms learned from experimental models are also involved in human disease. The sensitivity and specificity of anti-PLA2R autoantibodies in individuals with immunologically active idiopathic MN have also enabled the development of a serologic immunoassay for the noninvasive diagnosis of primary MN and monitoring of disease activity. It should be noted that, while the association of these anti-PLA2R autoantibodies with disease suggests a causal role, this has not yet been confirmed experimentally.
Thrombospondin type-1 domain-containing 7A — THSD7A is, like PLA2R, a transmembrane protein expressed on podocytes [40,41]. THSD7A may be the responsible antigen in approximately 10 percent of patients with idiopathic MN who are negative for anti-PLA2R antibodies. The association of THSD7A with MN was examined in a study of 154 patients with anti-PLA2R-negative idiopathic MN, 74 patients with anti-PLA2R-positive idiopathic MN, 76 patients with other glomerular disease, and 44 healthy controls [40]. Autoantibodies specific for THSD7A were identified in sera from 15 of 154 patients with anti-PLA2R-negative idiopathic MN but not in the sera from other individuals. In addition, the IgG that was eluted from the kidney biopsies of 1 of these 15 patients was specific for THSD7A, providing further support that THSD7A was the target antigen in these patients. THSD7A-associated MN has been found at a low frequency in North American and European cohorts, but it may be more prevalent in Japanese patients with primary MN [42].
THSD7A may also be involved in the pathogenesis of some cases of malignancy-associated MN [43]. (See 'Malignancy' below.)
Neutral endopeptidase — Neutral endopeptidase (NEP), which is expressed on podocytes, is the probable target in a rare antenatal form of MN [44,45]. The transplacental passage of anti-NEP antibodies (from mothers genetically deficient in NEP who were alloimmunized during a prior pregnancy) caused MN with subepithelial immune deposits (anti-NEP and NEP) in the fetus/neonate. Nephrotic syndrome resolved several months after birth, with disappearance of the deposits, upon clearance of the maternal antibodies. Although noncomplement-fixing IgG4 was the predominant IgG subclass of anti-NEP in alloimmunized NEP-deficient mothers, the development of proteinuria in their babies correlated with the additional presence of complement-fixing IgG1 anti-NEP [45]. This finding, together with reports showing that C5b-9 is shed into the urine of patients with recent-onset MN [46,47], provides additional evidence that the observations in Heymann nephritis are relevant to the human disease. It has yet to be determined, however, whether the IgG4 anti-PLA2R antibodies that predominate in idiopathic MN or accompanying IgG1 antibodies are responsible for complement activation.
Intracellular antigens — In addition to the PLA2R, THSD7A, and NEP, antibodies directed against other antigens expressed by podocytes may contribute to the pathogenesis of MN [48-50]. In one study, for example, serum IgG4 reactivity against aldose reductase, superoxide dismutase 2, and alpha-enolase, as well as the PLA2R and neutral endopeptidase, was measured in 186 patients with MN, 36 patients with focal glomerulosclerosis, and 60 patients with immunoglobulin A (IgA) nephropathy [48]. Elevated titers of IgG4 against the PLA2R, alpha-enolase, aldose reductase, and superoxide dismutase 2 were found in 60, 43, 34, and 28 percent of patients with MN, respectively, but not in patients with other glomerular diseases. Approximately one-half of the patients who were negative for antibodies against the PLA2R had an elevated titer for one of the other three antibodies. Although these antigens are predominantly intracellular, it has been proposed that podocyte injury causes the intracellular enzymes to translocate to the cell surface where they are accessible to the circulating antibodies, causing amplification of the immune injury and possibly aggravating the course of the disease.
Cationic bovine serum albumin — Antibodies to a cationic form of bovine serum albumin (BSA) are present in a small number of children with MN [51]. The BSA antigen, which was found within the immune deposits of biopsy specimens from these patients, is thought to be absorbed from the relatively underdeveloped pediatric intestinal tract in a partially digested or undigested form and then serve as a planted antigen within the glomerular capillary wall. Antibodies reactive with bovine, but not human, serum albumin were eluted from the kidney biopsy specimen in one case.
Other antigens — Other antigens have been identified within the glomerular immune deposits in patients with secondary MN [52]. These antigens include double-stranded DNA in systemic lupus erythematosus (SLE), thyroglobulin in thyroiditis, hepatitis B antigen, treponemal antigen and Helicobacter pylori in the relevant infections, and carcinoembryonic antigen and prostate specific antigen in malignancy. The pathogenicity of these antigens is unproven.
Role of T cells — T helper cells activate different immune effector mechanisms and appear to play a role in the pathogenesis of glomerulonephritis and may also participate in the genesis of proteinuria in MN. The T helper subset Th1 tends to predominate in proliferative and crescentic forms of glomerulonephritis, whereas Th2 predominates in MN and minimal change disease [53,54]. (See "The adaptive cellular immune response", section on 'Cytokine secretion profiles of activated T cells' and "Mechanisms of immune injury of the glomerulus", section on 'T cells'.)
In support of a pathogenetic role for Th2 in MN is the observation in a model of lupus nephritis that deletion of the WSX-1 gene (encoding a cytokine receptor integral for mounting a Th1 response) causes a shift toward a Th2 response and converts the diffuse proliferative pattern that is typically seen to a membranous pattern [55]. The site of the target antigen and subepithelial location of the immune deposits also favor antibody- and complement-mediated injury to the podocyte, rather than a direct cellular inflammatory lesion (which is typically associated with subendothelial immune deposits and glomerular endothelial injury) [56,57].
Genetics
Associated Conditions
Gross Pathology
Microscopic Pathology
References
-
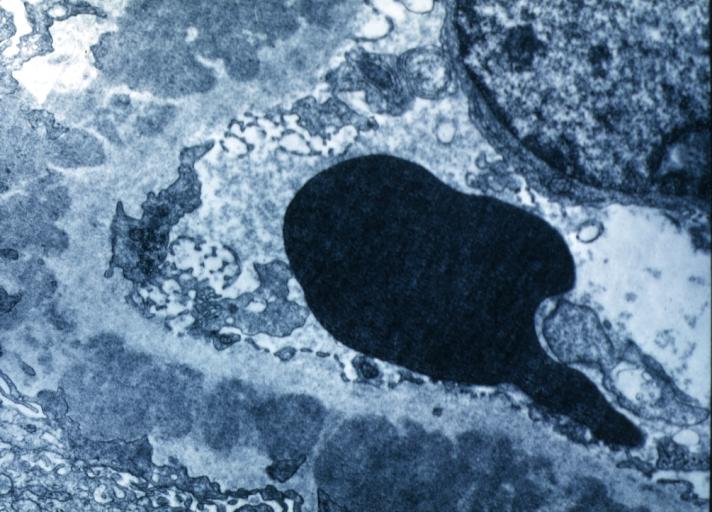
Membranous Glomerulonephritis: Electron micrography. An excellent example to show thickened basement membrane and immune complexes.
-
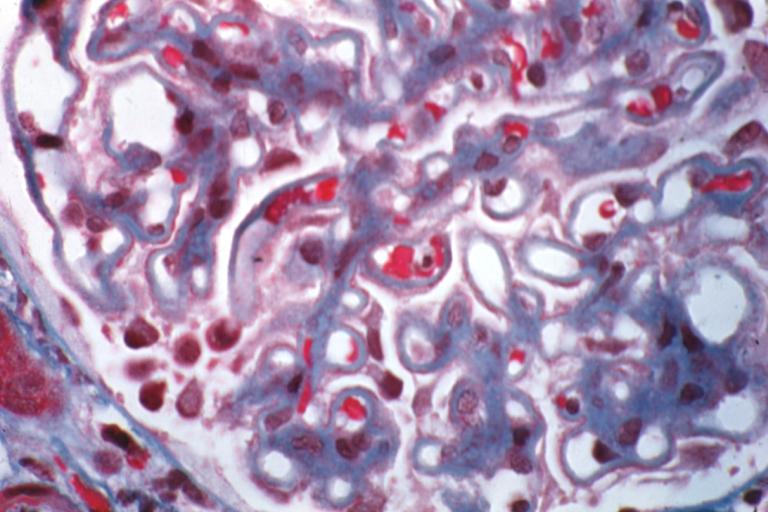
Membranous Glomerulonephritis: Micro trichrome high mag excellent to show thickened capillary basement membranes
-
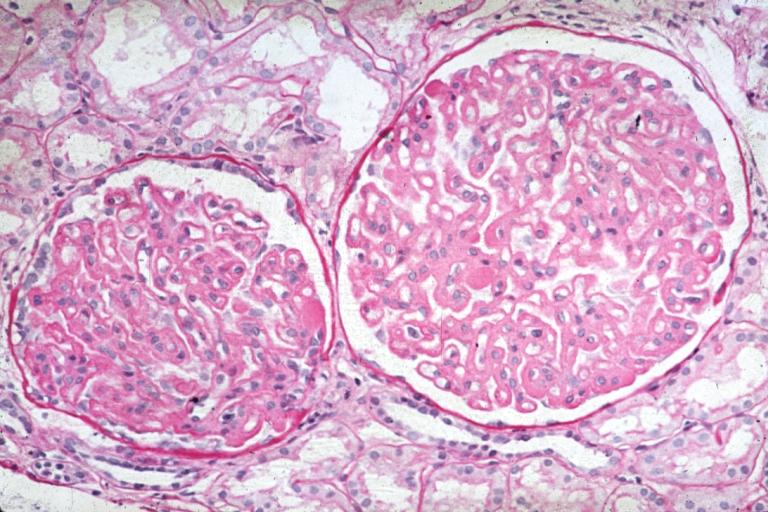
Membranous Glomerulonephritis: Micro PAS high mag excellent example of this lesion
-
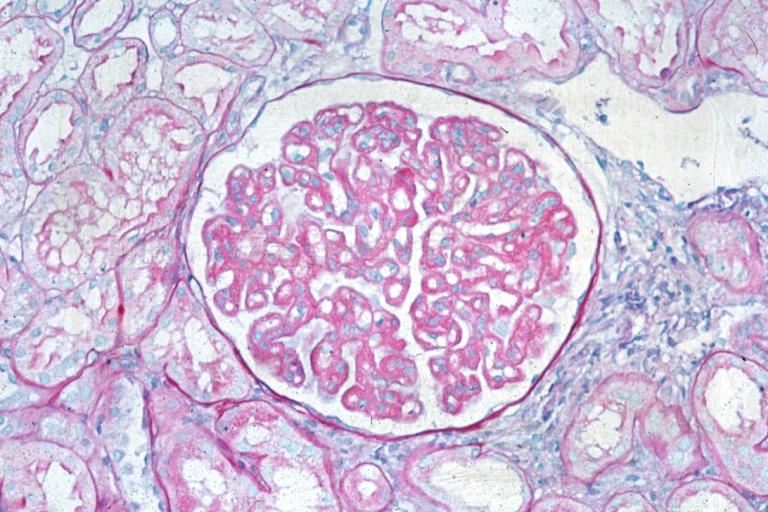
Membranous Glomerulonephritis: Micro PAS med mag



