Axitinib: Difference between revisions
No edit summary |
No edit summary |
||
| Line 3: | Line 3: | ||
|genericName=Axitinib | |genericName=Axitinib | ||
|aOrAn=an | |aOrAn=an | ||
|drugClass=antineoplastic agent | |drugClass=antineoplastic agent | ||
|indicationType=treatment | |indicationType=treatment | ||
|indication=advanced [[renal cell carcinoma]] (RCC) after failure of one prior systemic therapy. | |indication=advanced [[renal cell carcinoma]] (RCC) after failure of one prior systemic therapy. | ||
| Line 148: | Line 148: | ||
<!--Clinical Trials Experience--> | <!--Clinical Trials Experience--> | ||
|clinicalTrials= | |clinicalTrials=Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice. | ||
The safety of INLYTA has been evaluated in 715 patients in monotherapy studies, which included 537 patients with advanced RCC. The data described [see ADVERSE REACTIONS (6.1)] reflect exposure to INLYTA in 359 patients with advanced RCC who participated in a randomized clinical study versus sorafenib [see CLINICAL STUDIES (14)]. | |||
The following risks, including appropriate action to be taken, are discussed in greater detail in other sections of the label [see WARNINGS AND PRECAUTIONS (5.1–5.13)]: hypertension, arterial thromboembolic events, venous thromboembolic events, hemorrhage, cardiac failure, gastrointestinal perforation and fistula formation, thyroid dysfunction, wound healing complications, RPLS, proteinuria, elevation of liver enzymes, hepatic impairment and fetal development. | |||
6.1 Clinical Trials Experience | |||
The median duration of treatment was 6.4 months (range 0.03 to 22.0) for patients who received INLYTA and 5.0 months (range 0.03 to 20.1) for patients who received sorafenib. Dose modifications or temporary delay of treatment due to an adverse reaction occurred in 199/359 patients (55%) receiving INLYTA and 220/355 patients (62%) receiving sorafenib. Permanent discontinuation due to an adverse reaction occurred in 34/359 patients (9%) receiving INLYTA and 46/355 patients (13%) receiving sorafenib. | |||
The most common (≥20%) adverse reactions observed following treatment with INLYTA were diarrhea, hypertension, fatigue, decreased appetite, nausea, dysphonia, palmar-plantar erythrodysesthesia (hand-foot) syndrome, weight decreased, vomiting, asthenia, and constipation. Table 1 presents adverse reactions reported in ≥10% patients who received INLYTA or sorafenib. | |||
table1 | |||
Selected adverse reactions (all grades) that were reported in <10% of patients treated with INLYTA included dizziness (9%), upper abdominal pain (8%), myalgia (7%), dehydration (6%), epistaxis (6%), anemia (4%), hemorrhoids (4%), hematuria (3%), tinnitus (3%), lipase increased (3%), glossodynia (3%), pulmonary embolism (2%), rectal hemorrhage (2%), hemoptysis (2%), deep vein thrombosis (1%), retinal-vein occlusion/thrombosis (1%), polycythemia (1%), and transient ischemic attack (1%). | |||
Table 2 presents the most common laboratory abnormalities reported in ≥10% patients who received INLYTA or sorafenib. | |||
table2 | |||
Selected laboratory abnormalities (all grades) that were reported in <10% of patients treated with INLYTA included hemoglobin increased (above the upper limit of normal) (9% for INLYTA versus 1% for sorafenib) and hypercalcemia (6% for INLYTA versus 2% for sorafenib). | |||
|postmarketing=There is limited information regarding <i>Postmarketing Experience</i> of {{PAGENAME}} in the drug label. | |postmarketing=There is limited information regarding <i>Postmarketing Experience</i> of {{PAGENAME}} in the drug label. | ||
| Line 270: | Line 220: | ||
<!--Drug Interactions--> | <!--Drug Interactions--> | ||
|drugInteractions=* | |drugInteractions=* In vitro data indicate that axitinib is metabolized primarily by CYP3A4/5 and, to a lesser extent, CYP1A2, CYP2C19, and uridine diphosphate-glucuronosyltransferase (UGT) 1A1. | ||
=====CYP3A4/5 Inhibitors===== | |||
*Co-administration of ketoconazole, a strong inhibitor of CYP3A4/5, increased the plasma exposure of axitinib in healthy volunteers. Co-administration of INLYTA with strong CYP3A4/5 inhibitors should be avoided. Grapefruit or grapefruit juice may also increase axitinib plasma concentrations and should be avoided. Selection of concomitant medication with no or minimal CYP3A4/5 inhibition potential is recommended. If a strong CYP3A4/5 inhibitor must be co-administered, the INLYTA dose should be reduced [see DOSAGE AND ADMINISTRATION (2.2) and CLINICAL PHARMACOLOGY (12.3)]. | |||
=====CYP3A4/5 Inducers===== | |||
*Co-administration of rifampin, a strong inducer of CYP3A4/5, reduced the plasma exposure of axitinib in healthy volunteers. Co-administration of INLYTA with strong CYP3A4/5 inducers (e.g., rifampin, dexamethasone, phenytoin, carbamazepine, rifabutin, rifapentin, phenobarbital, and St. John's wort) should be avoided. Selection of concomitant medication with no or minimal CYP3A4/5 induction potential is recommended [see DOSAGE AND ADMINISTRATION (2.2) and CLINICAL PHARMACOLOGY (12.3)]. Moderate CYP3A4/5 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, and nafcillin) may also reduce the plasma exposure of axitinib and should be avoided if possible. | |||
<!--Use in Specific Populations--> | <!--Use in Specific Populations--> | ||
|useInPregnancyFDA=* | |FDAPregCat=D | ||
|useInPregnancyFDA=* INLYTA can cause fetal harm when administered to a pregnant woman based on its mechanism of action. There are no adequate and well-controlled studies in pregnant women using INLYTA. In developmental toxicity studies in mice, axitinib was teratogenic, embryotoxic and fetotoxic at maternal exposures that were lower than human exposures at the recommended clinical dose. | |||
*Women of childbearing potential should be advised to avoid becoming pregnant while receiving INLYTA. If this drug is used during pregnancy, or if a patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus | |||
Pregnancy Category D [see WARNINGS AND PRECAUTIONS (5.13)]. | |||
There are no adequate and well-controlled studies with INLYTA in pregnant women. INLYTA can cause fetal harm when administered to a pregnant woman based on its mechanism of action. Axitinib was teratogenic, embryotoxic and fetotoxic in mice at exposures lower than human exposures at the recommended starting dose. If this drug is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. | |||
Oral axitinib administered twice daily to female mice prior to mating and through the first week of pregnancy caused an increase in post-implantation loss at all doses tested (≥15 mg/kg/dose, approximately 10 times the systemic exposure (AUC) in patients at the recommended starting dose). In an embryo-fetal developmental toxicity study, pregnant mice received oral doses of 0.15, 0.5 and 1.5 mg/kg/dose axitinib twice daily during the period of organogenesis. Embryo-fetal toxicities observed in the absence of maternal toxicity included malformation (cleft palate) at 1.5 mg/kg/dose (approximately 0.5 times the AUC in patients at the recommended starting dose) and variation in skeletal ossification at ≥0.5 mg/kg/dose (approximately 0.15 times the AUC in patients at the recommended starting dose). | |||
|useInPregnancyAUS=* '''Australian Drug Evaluation Committee (ADEC) Pregnancy Category''' | |useInPregnancyAUS=* '''Australian Drug Evaluation Committee (ADEC) Pregnancy Category''' | ||
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of {{PAGENAME}} in women who are pregnant. | There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of {{PAGENAME}} in women who are pregnant. | ||
|useInLaborDelivery=There is no FDA guidance on use of {{PAGENAME}} during labor and delivery. | |useInLaborDelivery=There is no FDA guidance on use of {{PAGENAME}} during labor and delivery. | ||
|useInNursing= | |useInNursing=*It is not known whether axitinib is excreted in human milk. *Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from INLYTA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. | ||
|useInPed= | |useInPed=*The safety and efficacy of INLYTA in pediatric patients have not been studied. | ||
|useInGeri= | *Toxicities in bone and teeth were observed in immature mice and dogs administered oral axitinib twice daily for 1 month or longer. Effects in bone consisted of thickened growth plates in mice and dogs at ≥15 mg/kg/dose (approximately 6 and 15 times, respectively, the systemic exposure (AUC) in patients at the recommended starting dose). Abnormalities in growing incisor teeth (including dental caries, malocclusions and broken and/or missing teeth) were observed in mice administered oral axitinib twice daily at ≥5 mg/kg/dose (approximately 1.5 times the AUC in patients at the recommended starting dose). Other toxicities of potential concern to pediatric patients have not been evaluated in juvenile animals. | ||
|useInGeri=*In a controlled clinical study with INLYTA for the treatment of patients with RCC, 123/359 patients (34%) treated with INLYTA were ≥65 years of age. Although greater sensitivity in some older individuals cannot be ruled out, no overall differences were observed in the safety and effectiveness of INLYTA between patients who were ≥65 years of age and younger. | |||
*No dosage adjustment is required in elderly patients | |||
|useInGender=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific gender populations. | |useInGender=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific gender populations. | ||
|useInRace=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific racial populations. | |useInRace=There is no FDA guidance on the use of {{PAGENAME}} with respect to specific racial populations. | ||
|useInRenalImpair= | |useInRenalImpair=*No dedicated renal impairment trial for axitinib has been conducted. Based on the population pharmacokinetic analyses, no significant difference in axitinib clearance was observed in patients with pre-existing mild to severe renal impairment (15 mL/min ≤creatinine clearance [CLcr] <89 mL/min) [see CLINICAL PHARMACOLOGY (12.3)]. No starting dose adjustment is needed for patients with pre-existing mild to severe renal impairment. Caution should be used in patients with end-stage renal disease (CLcr <15 mL/min).. | ||
|useInHepaticImpair= | |useInHepaticImpair=*In a dedicated hepatic impairment trial, compared to subjects with normal hepatic function, systemic exposure following a single dose of INLYTA was similar in subjects with baseline mild hepatic impairment (Child-Pugh class A) and higher in subjects with baseline moderate hepatic impairment (Child-Pugh class B). | ||
*No starting dose adjustment is required when administering INLYTA to patients with mild hepatic impairment (Child-Pugh class A). A starting dose decrease is recommended when administering INLYTA to patients with moderate hepatic impairment (Child-Pugh class B) [see DOSAGE AND ADMINISTRATION (2.2), WARNINGS AND PRECAUTIONS (5.12), and CLINICAL PHARMACOLOGY (12.3)]. | |||
*INLYTA has not been studied in subjects with severe hepatic impairment (Child-Pugh class C). | |||
|useInReproPotential=There is no FDA guidance on the use of {{PAGENAME}} in women of reproductive potentials and males. | |useInReproPotential=There is no FDA guidance on the use of {{PAGENAME}} in women of reproductive potentials and males. | ||
|useInImmunocomp=There is no FDA guidance one the use of {{PAGENAME}} in patients who are immunocompromised. | |useInImmunocomp=There is no FDA guidance one the use of {{PAGENAME}} in patients who are immunocompromised. | ||
Revision as of 14:57, 30 January 2015
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Aparna Vuppala, M.B.B.S. [2]
Disclaimer
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Axitinib is an antineoplastic agent that is FDA approved for the treatment of advanced renal cell carcinoma (RCC) after failure of one prior systemic therapy.. Common adverse reactions include .
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Renal cell carcinoma (RCC)
- INLYTA is indicated for the treatment of advanced renal cell carcinoma (RCC) after failure of one prior systemic therapy.
Recommended Dosing
- The recommended starting oral dose of INLYTA is 5 mg twice daily. Administer INLYTA doses approximately 12 hours apart with or without food [see CLINICAL PHARMACOLOGY (12.3)]. INLYTA should be swallowed whole with a glass of water.
- If the patient vomits or misses a dose, an additional dose should not be taken. The next prescribed dose should be taken at the usual time.
Dose Modification Guidelines
- Dose increase or reduction is recommended based on individual safety and tolerability.
- Over the course of treatment, patients who tolerate INLYTA for at least two consecutive weeks with no adverse reactions >Grade 2 (according to the Common Toxicity Criteria for Adverse Events [CTCAE]), are normotensive, and are not receiving anti-hypertension medication, may have their dose increased. When a dose increase from 5 mg twice daily is recommended, the INLYTA dose may be increased to 7 mg twice daily, and further to 10 mg twice daily using the same criteria.
- Over the course of treatment, management of some adverse drug reactions may require temporary interruption or permanent discontinuation and/or dose reduction of INLYTA therapy [see WARNINGS AND PRECAUTIONS (5)]. If dose reduction from 5 mg twice daily is required, the recommended dose is 3 mg twice daily. If additional dose reduction is required, the recommended dose is 2 mg twice daily.
- Strong CYP3A4/5 Inhibitors: The concomitant use of strong CYP3A4/5 inhibitors should be avoided (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole). Selection of an alternate concomitant medication with no or minimal CYP3A4/5 inhibition potential is recommended. *Although INLYTA dose adjustment has not been studied in patients receiving strong CYP3A4/5 inhibitors, if a strong CYP3A4/5 inhibitor must be co-administered, a dose decrease of INLYTA by approximately half is recommended, as this dose reduction is predicted to adjust the axitinib area under the plasma concentration vs time curve (AUC) to the range observed without inhibitors. The subsequent doses can be increased or decreased based on individual safety and tolerability. If co-administration of the strong inhibitor is discontinued, the INLYTA dose should be returned (after 3 – 5 half-lives of the inhibitor) to that used prior to initiation of the strong CYP3A4/5 inhibitor [see DRUG INTERACTIONS (7.1) and CLINICAL PHARMACOLOGY (12.3)].
- Hepatic Impairment: No starting dose adjustment is required when administering INLYTA to patients with mild hepatic impairment (Child-Pugh class A). Based on the pharmacokinetic data, the INLYTA starting dose should be reduced by approximately half in patients with baseline moderate hepatic impairment (Child-Pugh class B). The subsequent doses can be increased or decreased based on individual safety and tolerability. INLYTA has not been studied in patients with severe hepatic impairment (Child-Pugh class C)
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
Condition1
- Developed by:
- Class of Recommendation:
- Strength of Evidence:
- Dosing Information
- Dosage
Condition2
There is limited information regarding Off-Label Guideline-Supported Use of Axitinib in adult patients.
Non–Guideline-Supported Use
Condition1
- Dosing Information
- Dosage
Condition2
There is limited information regarding Off-Label Non–Guideline-Supported Use of Axitinib in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
Condition1
- Dosing Information
- Dosage
Condition2
There is limited information regarding FDA-Labeled Use of Axitinib in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
Condition1
- Developed by:
- Class of Recommendation:
- Strength of Evidence:
- Dosing Information
- Dosage
Condition2
There is limited information regarding Off-Label Guideline-Supported Use of Axitinib in pediatric patients.
Non–Guideline-Supported Use
Condition1
- Dosing Information
- Dosage
Condition2
There is limited information regarding Off-Label Non–Guideline-Supported Use of Axitinib in pediatric patients.
Contraindications
- None
Warnings
Hypertension and Hypertensive Crisis
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, hypertension was reported in 145/359 patients (40%) receiving INLYTA and 103/355 patients (29%) receiving sorafenib. Grade 3/4 hypertension was observed in 56/359 patients (16%) receiving INLYTA and 39/355 patients (11%) receiving sorafenib. Hypertensive crisis was reported in 2/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib. The median onset time for hypertension (systolic blood pressure >150 mmHg or diastolic blood pressure >100 mmHg) was within the first month of the start of INLYTA treatment and blood pressure increases have been observed as early as 4 days after starting INLYTA. Hypertension was managed with standard antihypertensive therapy. Discontinuation of INLYTA treatment due to hypertension occurred in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib [see ADVERSE REACTIONS (6.1)].
- Blood pressure should be well-controlled prior to initiating INLYTA. Patients should be monitored for hypertension and treated as needed with standard anti-hypertensive therapy. In the case of persistent hypertension despite use of anti-hypertensive medications, reduce the INLYTA dose. Discontinue INLYTA if hypertension is severe and persistent despite anti-hypertensive therapy and dose reduction of INLYTA, and discontinuation should be considered if there is evidence of hypertensive crisis. If INLYTA is interrupted, patients receiving antihypertensive medications should be monitored for hypotension [see DOSAGE AND ADMINISTRATION (2.2)].
Arterial Thromboembolic Events
- In clinical trials, arterial thromboembolic events have been reported, including deaths. In a controlled clinical study with INLYTA for the treatment of patients with RCC, Grade 3/4 arterial thromboembolic events were reported in 4/359 patients (1%) receiving INLYTA and 4/355 patients (1%) receiving sorafenib. Fatal cerebrovascular accident was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib [see ADVERSE REACTIONS (6.1)].
- In clinical trials with INLYTA, arterial thromboembolic events (including transient ischemic attack, cerebrovascular accident, myocardial infarction, and retinal artery occlusion) were reported in 17/715 patients (2%), with two deaths secondary to cerebrovascular accident.
- Use INLYTA with caution in patients who are at risk for, or who have a history of, these events. INLYTA has not been studied in patients who had an arterial thromboembolic event within the previous 12 months.
Venous Thromboembolic Events
- In clinical trials, venous thromboembolic events have been reported, including deaths. In a controlled clinical study with INLYTA for the treatment of patients with RCC, venous thromboembolic events were reported in 11/359 patients (3%) receiving INLYTA and 2/355 patients (1%) receiving sorafenib. Grade 3/4 venous thromboembolic events were reported in 9/359 patients (3%) receiving INLYTA (including pulmonary embolism, deep vein thrombosis, retinal vein occlusion and retinal vein thrombosis) and 2/355 patients (1%) receiving sorafenib. Fatal pulmonary embolism was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib. In clinical trials with INLYTA, venous thromboembolic events were reported in 22/715 patients (3%), with two deaths secondary to pulmonary embolism.
- Use INLYTA with caution in patients who are at risk for, or who have a history of, these events. INLYTA has not been studied in patients who had a venous thromboembolic event within the previous 6 months.
Hemorrhage
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, hemorrhagic events were reported in 58/359 patients (16%) receiving INLYTA and 64/355 patients (18%) receiving sorafenib. Grade 3/4 hemorrhagic events were reported in 5/359 (1%) patients receiving INLYTA (including cerebral hemorrhage, hematuria, hemoptysis, lower gastrointestinal hemorrhage, and melena) and 11/355 (3%) patients receiving sorafenib. Fatal hemorrhage was reported in 1/359 patients (<1%) receiving INLYTA (gastric hemorrhage) and 3/355 patients (1%) receiving sorafenib.
- INLYTA has not been studied in patients who have evidence of untreated brain metastasis or recent active gastrointestinal bleeding and should not be used in those patients. If any bleeding requires medical intervention, temporarily interrupt the INLYTA dose.
Cardiac Failure
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, cardiac failure was reported in 6/359 patients (2%) receiving INLYTA and 3/355 patients (1%) receiving sorafenib. Grade 3/4 cardiac failure was observed in 2/359 patients (1%) receiving INLYTA and 1/355 patients (<1%) receiving sorafenib. *Fatal cardiac failure was reported in 2/359 patients (1%) receiving INLYTA and 1/355 patients (<1%) receiving sorafenib. Monitor for signs or symptoms of cardiac failure throughout treatment with INLYTA. Management of cardiac failure may require permanent discontinuation of INLYTA.
Gastrointestinal Perforation and Fistula Formation
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, gastrointestinal perforation was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib. In clinical trials with INLYTA, gastrointestinal perforation was reported in 5/715 patients (1%), including one death. In addition to cases of gastrointestinal perforation, fistulas were reported in 4/715 patients (1%).
- Monitor for symptoms of gastrointestinal perforation or fistula periodically throughout treatment with INLYTA.
Thyroid Dysfunction
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, hypothyroidism was reported in 69/359 patients (19%) receiving INLYTA and 29/355 patients (8%) receiving sorafenib. Hyperthyroidism was reported in 4/359 patients (1%) receiving INLYTA and 4/355 patients (1%) receiving sorafenib. In patients who had thyroid stimulating hormone (TSH) <5 μU/mL before treatment, elevations of TSH to ≥10 μU/mL occurred in 79/245 patients (32%) receiving INLYTA and 25/232 patients (11%) receiving sorafenib [see ADVERSE REACTIONS (6.1)].
- Monitor thyroid function before initiation of, and periodically throughout, treatment with INLYTA. Treat hypothyroidism and hyperthyroidism according to standard medical practice to maintain euthyroid state.
Wound Healing Complications
- No formal studies of the effect of INLYTA on wound healing have been conducted.
- Stop treatment with INLYTA at least 24 hours prior to scheduled surgery. The decision to resume INLYTA therapy after surgery should be based on clinical judgment of adequate wound healing.
Reversible Posterior Leukoencephalopathy Syndrome
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, reversible posterior leukoencephalopathy syndrome (RPLS) was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib [see ADVERSE REACTIONS (6.1)]. There were two additional reports of RPLS in other clinical trials with INLYTA.
- RPLS is a neurological disorder which can present with headache, seizure, lethargy, confusion, blindness and other visual and neurologic disturbances. Mild to severe hypertension may be present. Magnetic resonance imaging is necessary to confirm the diagnosis of RPLS. Discontinue INLYTA in patients developing RPLS. *The safety of reinitiating INLYTA therapy in patients previously experiencing RPLS is not known.
Proteinuria
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, proteinuria was reported in 39/359 patients (11%) receiving INLYTA and 26/355 patients (7%) receiving sorafenib. Grade 3 proteinuria was reported in 11/359 patients (3%) receiving INLYTA and 6/355 patients (2%) receiving sorafenib [see ADVERSE REACTIONS (6.1)].
- Monitoring for proteinuria before initiation of, and periodically throughout, treatment with INLYTA is recommended. For patients who develop moderate to severe proteinuria, reduce the dose or temporarily interrupt INLYTA treatment.
Elevation of Liver Enzymes
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, alanine aminotransferase (ALT) elevations of all grades occurred in 22% of patients on both arms, with Grade 3/4 events in <1% of patients on the INLYTA arm and 2% of patients on the sorafenib arm.
- Monitor ALT, aspartate aminotransferase (AST) and bilirubin before initiation of and periodically throughout treatment with INLYTA.
Hepatic Impairment
- The systemic exposure to axitinib was higher in subjects with moderate hepatic impairment (Child-Pugh class B) compared to subjects with normal hepatic function. A dose decrease is recommended when administering INLYTA to patients with moderate hepatic impairment (Child-Pugh class B). INLYTA has not been studied in patients with severe hepatic impairment (Child-Pugh class C) [see DOSAGE AND ADMINISTRATION (2.2), USE IN SPECIFIC POPULATIONS (8.6), and CLINICAL PHARMACOLOGY (12.3)].
Precautions
- Description
Adverse Reactions
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of INLYTA has been evaluated in 715 patients in monotherapy studies, which included 537 patients with advanced RCC. The data described [see ADVERSE REACTIONS (6.1)] reflect exposure to INLYTA in 359 patients with advanced RCC who participated in a randomized clinical study versus sorafenib [see CLINICAL STUDIES (14)].
The following risks, including appropriate action to be taken, are discussed in greater detail in other sections of the label [see WARNINGS AND PRECAUTIONS (5.1–5.13)]: hypertension, arterial thromboembolic events, venous thromboembolic events, hemorrhage, cardiac failure, gastrointestinal perforation and fistula formation, thyroid dysfunction, wound healing complications, RPLS, proteinuria, elevation of liver enzymes, hepatic impairment and fetal development.
6.1 Clinical Trials Experience The median duration of treatment was 6.4 months (range 0.03 to 22.0) for patients who received INLYTA and 5.0 months (range 0.03 to 20.1) for patients who received sorafenib. Dose modifications or temporary delay of treatment due to an adverse reaction occurred in 199/359 patients (55%) receiving INLYTA and 220/355 patients (62%) receiving sorafenib. Permanent discontinuation due to an adverse reaction occurred in 34/359 patients (9%) receiving INLYTA and 46/355 patients (13%) receiving sorafenib.
The most common (≥20%) adverse reactions observed following treatment with INLYTA were diarrhea, hypertension, fatigue, decreased appetite, nausea, dysphonia, palmar-plantar erythrodysesthesia (hand-foot) syndrome, weight decreased, vomiting, asthenia, and constipation. Table 1 presents adverse reactions reported in ≥10% patients who received INLYTA or sorafenib. table1 Selected adverse reactions (all grades) that were reported in <10% of patients treated with INLYTA included dizziness (9%), upper abdominal pain (8%), myalgia (7%), dehydration (6%), epistaxis (6%), anemia (4%), hemorrhoids (4%), hematuria (3%), tinnitus (3%), lipase increased (3%), glossodynia (3%), pulmonary embolism (2%), rectal hemorrhage (2%), hemoptysis (2%), deep vein thrombosis (1%), retinal-vein occlusion/thrombosis (1%), polycythemia (1%), and transient ischemic attack (1%).
Table 2 presents the most common laboratory abnormalities reported in ≥10% patients who received INLYTA or sorafenib. table2 Selected laboratory abnormalities (all grades) that were reported in <10% of patients treated with INLYTA included hemoglobin increased (above the upper limit of normal) (9% for INLYTA versus 1% for sorafenib) and hypercalcemia (6% for INLYTA versus 2% for sorafenib).
Postmarketing Experience
There is limited information regarding Postmarketing Experience of Axitinib in the drug label.
Body as a Whole
Cardiovascular
Digestive
Endocrine
Hematologic and Lymphatic
Metabolic and Nutritional
Musculoskeletal
Neurologic
Respiratory
Skin and Hypersensitivy Reactions
Special Senses
Urogenital
Miscellaneous
Drug Interactions
- In vitro data indicate that axitinib is metabolized primarily by CYP3A4/5 and, to a lesser extent, CYP1A2, CYP2C19, and uridine diphosphate-glucuronosyltransferase (UGT) 1A1.
CYP3A4/5 Inhibitors
- Co-administration of ketoconazole, a strong inhibitor of CYP3A4/5, increased the plasma exposure of axitinib in healthy volunteers. Co-administration of INLYTA with strong CYP3A4/5 inhibitors should be avoided. Grapefruit or grapefruit juice may also increase axitinib plasma concentrations and should be avoided. Selection of concomitant medication with no or minimal CYP3A4/5 inhibition potential is recommended. If a strong CYP3A4/5 inhibitor must be co-administered, the INLYTA dose should be reduced [see DOSAGE AND ADMINISTRATION (2.2) and CLINICAL PHARMACOLOGY (12.3)].
CYP3A4/5 Inducers
- Co-administration of rifampin, a strong inducer of CYP3A4/5, reduced the plasma exposure of axitinib in healthy volunteers. Co-administration of INLYTA with strong CYP3A4/5 inducers (e.g., rifampin, dexamethasone, phenytoin, carbamazepine, rifabutin, rifapentin, phenobarbital, and St. John's wort) should be avoided. Selection of concomitant medication with no or minimal CYP3A4/5 induction potential is recommended [see DOSAGE AND ADMINISTRATION (2.2) and CLINICAL PHARMACOLOGY (12.3)]. Moderate CYP3A4/5 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, and nafcillin) may also reduce the plasma exposure of axitinib and should be avoided if possible.
Use in Specific Populations
Pregnancy
- INLYTA can cause fetal harm when administered to a pregnant woman based on its mechanism of action. There are no adequate and well-controlled studies in pregnant women using INLYTA. In developmental toxicity studies in mice, axitinib was teratogenic, embryotoxic and fetotoxic at maternal exposures that were lower than human exposures at the recommended clinical dose.
- Women of childbearing potential should be advised to avoid becoming pregnant while receiving INLYTA. If this drug is used during pregnancy, or if a patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus
Pregnancy Category D [see WARNINGS AND PRECAUTIONS (5.13)].
There are no adequate and well-controlled studies with INLYTA in pregnant women. INLYTA can cause fetal harm when administered to a pregnant woman based on its mechanism of action. Axitinib was teratogenic, embryotoxic and fetotoxic in mice at exposures lower than human exposures at the recommended starting dose. If this drug is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus.
Oral axitinib administered twice daily to female mice prior to mating and through the first week of pregnancy caused an increase in post-implantation loss at all doses tested (≥15 mg/kg/dose, approximately 10 times the systemic exposure (AUC) in patients at the recommended starting dose). In an embryo-fetal developmental toxicity study, pregnant mice received oral doses of 0.15, 0.5 and 1.5 mg/kg/dose axitinib twice daily during the period of organogenesis. Embryo-fetal toxicities observed in the absence of maternal toxicity included malformation (cleft palate) at 1.5 mg/kg/dose (approximately 0.5 times the AUC in patients at the recommended starting dose) and variation in skeletal ossification at ≥0.5 mg/kg/dose (approximately 0.15 times the AUC in patients at the recommended starting dose).
Pregnancy Category (AUS):
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Axitinib in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Axitinib during labor and delivery.
Nursing Mothers
- It is not known whether axitinib is excreted in human milk. *Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from INLYTA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
- The safety and efficacy of INLYTA in pediatric patients have not been studied.
- Toxicities in bone and teeth were observed in immature mice and dogs administered oral axitinib twice daily for 1 month or longer. Effects in bone consisted of thickened growth plates in mice and dogs at ≥15 mg/kg/dose (approximately 6 and 15 times, respectively, the systemic exposure (AUC) in patients at the recommended starting dose). Abnormalities in growing incisor teeth (including dental caries, malocclusions and broken and/or missing teeth) were observed in mice administered oral axitinib twice daily at ≥5 mg/kg/dose (approximately 1.5 times the AUC in patients at the recommended starting dose). Other toxicities of potential concern to pediatric patients have not been evaluated in juvenile animals.
Geriatic Use
- In a controlled clinical study with INLYTA for the treatment of patients with RCC, 123/359 patients (34%) treated with INLYTA were ≥65 years of age. Although greater sensitivity in some older individuals cannot be ruled out, no overall differences were observed in the safety and effectiveness of INLYTA between patients who were ≥65 years of age and younger.
- No dosage adjustment is required in elderly patients
Gender
There is no FDA guidance on the use of Axitinib with respect to specific gender populations.
Race
There is no FDA guidance on the use of Axitinib with respect to specific racial populations.
Renal Impairment
- No dedicated renal impairment trial for axitinib has been conducted. Based on the population pharmacokinetic analyses, no significant difference in axitinib clearance was observed in patients with pre-existing mild to severe renal impairment (15 mL/min ≤creatinine clearance [CLcr] <89 mL/min) [see CLINICAL PHARMACOLOGY (12.3)]. No starting dose adjustment is needed for patients with pre-existing mild to severe renal impairment. Caution should be used in patients with end-stage renal disease (CLcr <15 mL/min)..
Hepatic Impairment
- In a dedicated hepatic impairment trial, compared to subjects with normal hepatic function, systemic exposure following a single dose of INLYTA was similar in subjects with baseline mild hepatic impairment (Child-Pugh class A) and higher in subjects with baseline moderate hepatic impairment (Child-Pugh class B).
- No starting dose adjustment is required when administering INLYTA to patients with mild hepatic impairment (Child-Pugh class A). A starting dose decrease is recommended when administering INLYTA to patients with moderate hepatic impairment (Child-Pugh class B) [see DOSAGE AND ADMINISTRATION (2.2), WARNINGS AND PRECAUTIONS (5.12), and CLINICAL PHARMACOLOGY (12.3)].
- INLYTA has not been studied in subjects with severe hepatic impairment (Child-Pugh class C).
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Axitinib in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Axitinib in patients who are immunocompromised.
Administration and Monitoring
Administration
- Oral
- Intravenous
Monitoring
There is limited information regarding Monitoring of Axitinib in the drug label.
- Description
IV Compatibility
There is limited information regarding IV Compatibility of Axitinib in the drug label.
Overdosage
Acute Overdose
Signs and Symptoms
- Description
Management
- Description
Chronic Overdose
There is limited information regarding Chronic Overdose of Axitinib in the drug label.
Pharmacology
There is limited information regarding Axitinib Pharmacology in the drug label.
Mechanism of Action
Structure
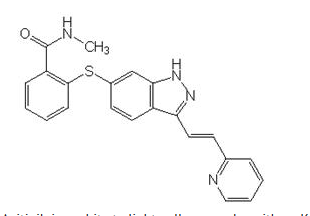
This image is provided by the National Library of Medicine.

Pharmacodynamics
There is limited information regarding Pharmacodynamics of Axitinib in the drug label.
Pharmacokinetics
There is limited information regarding Pharmacokinetics of Axitinib in the drug label.
Nonclinical Toxicology
There is limited information regarding Nonclinical Toxicology of Axitinib in the drug label.
Clinical Studies
There is limited information regarding Clinical Studies of Axitinib in the drug label.
How Supplied
Storage
There is limited information regarding Axitinib Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Axitinib |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Axitinib |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
There is limited information regarding Patient Counseling Information of Axitinib in the drug label.
Precautions with Alcohol
- Alcohol-Axitinib interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- ®[1]
Look-Alike Drug Names
- A® — B®[2]
Drug Shortage Status
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
- ↑ Empty citation (help)
- ↑ "http://www.ismp.org". External link in
|title=(help)
{{#subobject:
|Page Name=Axitinib
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Axitinib |Label Name=Axitinib11.png
}}
{{#subobject:
|Label Page=Axitinib |Label Name=Axitinib11.png
}}