Angiosarcoma
|
WikiDoc Resources for Angiosarcoma |
|
Articles |
|---|
|
Most recent articles on Angiosarcoma Most cited articles on Angiosarcoma |
|
Media |
|
Powerpoint slides on Angiosarcoma |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Angiosarcoma at Clinical Trials.gov Clinical Trials on Angiosarcoma at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Angiosarcoma
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Angiosarcoma Discussion groups on Angiosarcoma Patient Handouts on Angiosarcoma Directions to Hospitals Treating Angiosarcoma Risk calculators and risk factors for Angiosarcoma
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Angiosarcoma |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Shyam Patel [2]; Associate Editor(s)-in-Chief: Maria Fernanda Villarreal, M.D. [3]
Synonyms and keywords: Hemangiosarcoma; Pulmonary angiosarcoma; Vascular sarcoma
Overview
Angiosarcoma is a rare malignant vascular neoplasm of endothelial-type cells that line vessel walls. The peak age of incidence appears to be the 7th decade, and men are affected more than women. Angiosarcoma was first described by Dr. Juan Rosai, in 1976. The pathogenesis of angiosarcoma is characterized by a rapid and extensively infiltrating overgrowth of vascular epithelial cells. Angiosarcoma may arise in any part of the body, but is more common in soft tissue than in bone. Common angiosarcoma locations include the head and neck area, kidney, liver, lung, and and the most common site of radiation-induced angiosarcoma development is the breast. The PTPRB/PLCG1 genes are associated with the development of angiosarcoma; mutation of these genes result in aberrant angiogenesis. The imaging modality of choice for diagnosing angiosarcoma will depend on the location. For pulmonary angiosarcoma, the imaging modality of choice is enhanced CT scan. For other types angiosarcoma, the imaging modality of choice is MRI. On CT scan, findings suggestive of angiosarcoma may include vascular invasion, nodular enhancement (common), and a hypoattenuating mass. The mainstay adjuvant therapy for angiosarcoma is a doxorubicin-based regimen. The response rate for chemotherapy in patients with angiosarcoma is poor.
Historical Perspective
Angiosarcoma was first discovered by Dr. Juan Rosai, M.D. and colleagues in 1976.[1]
Classification
Angiosarcoma may be classified according to the clinical heterogeneity into two main groups, and every group can be subdivided into subtypes according to the anatomical location and etiology:[2] [3][4]
| Angiosarcoma | |
| Primary | Secondary |
| Cutaneous | Post Radiation Angiosarcoma |
| Breast | Lymphedema-associated Angiosarcoma |
| Soft tissue and Bone | Angiosarcoma due to exposure to mutatgens |
| Visceral | Visceral |
Staging
According to the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) and by Enneking classification, soft tissue sarcomas are classified to different stages based on the primary tumor characteristics, histological grading and the local or distant tumor involvement. Table below provides summarized information regarding staging of angiosarcoma:
| Stage | Grade | Site | Metastasis |
|---|---|---|---|
| Ia | Low grade (G1) | Intracompartmental (T1) | No metastasis (M0) |
| Ib | Low grade (G1) | Extracompartmental (T2) | No metastasis (M0) |
| IIa | High grade (G2) | Intracompartmental (T1) | No metastasis (M0) |
| IIb | High grade (G2) | Extracompartmental (T2) | No metastasis (M0) |
| IIIa | Low or High grade (G1-G2) | Intracompartmental (T1) | Metastasis (M1) |
| IIIb | Low or High grade (G1-G2) | Extracompartmental (T2) | Metastasis (M1) |
Pathophysiology
Pathogenesis
The pathogenesis of angiosarcoma is characterized by a rapid and extensive infiltrating overgrowth of vascular epithelial cells.[5] Angiosarcoma is a locally aggressive tumor with a high rate of lymph node infiltration and metastases.
Genetics
- Angiosarcomas demonstrate obvious unregulated vascular-specific receptor tyrosine kinases containing TIE1, KDR, TEK and FLT.The up-regulation of these genes and over-expression of vascular endothelial growth factor receptors can cause endothelial cell expansion, angiogenesis, and also vascular leaks in the structure of vessels. [6][7][8][9]
- KDR mutations are seen in primary breast angiosarcoma regardless of exposure to radiation.
- High level MYC amplification is seen is seen in radiation- induced and lymphedema-associated angiosarcoma.
- FLT4 amplification has been detected in 25% of secondary angiosarcomas.
Gross Pathology
On gross pathology, characteristic findings of angiosarcoma may include:[5]
- Red/dark tan lesion
- Typically poorly circumscribed
Microscopic Pathology
On microscopic histopathological analysis, characteristic findings of angiosarcoma may include:
- Spindle cell lesion
- Epitheloid lesion
- Numerous irrergular capillaries
- Appears red on low power
- Pleomorphic nuclei
- Hobnail morphology
- Numerous mitotic bodies
- Cytoplasmic vacuoles
- Luminal arrangement of cells
Causes
The most common cause of angiosarcoma appears to be therapeutic radiation, which was a well-recognized cause of hepatic angiosarcoma in the era when the thorium containing contrast agent Thorotrast was employed. Presently, the breast is the most common anatomic site affected by radiation-induced angiosarcoma. Angiosarcomas may arise after exposure to vinyl chloride, although they remain rare tumors even in an exposed population. Angiosarcomas are also observed after lymphedema from any cause, be it surgical, filarial, or congenital, and defined as Stewart-Treves syndrome. Common causes of angiosarcoma include:[5]
- Exposure to vinyl chloride monomer (VCM) for prolonged periods
- Exposure to polyvinyl chloride (PVC) polymerisation plants
- Exposure to arsenic-containing insecticides
- Previous exposure to thorium dioxide irradiation
Differentiating Angiosarcoma from Other Diseases
Angiosarcoma must be differentiated from other diseases that cause a highly vascular mass or non-healing cutaneous ulcerations such as:
Differentials for Cutaneous Angiosarcoma
Cutaneous angiosarcoma must be differentiated from other diseases with non-healing cutaneous ulcerations such as:[10][11][12]
- Basal cell carcinoma
- Keratoacanthoma
- Nodular melanoma
- Mycosis fungoides
- Kaposi sarcoma
- Kaposi-like hemangioendothelioma
- Angiolymphoid hyperplasia
Differentials for Non-cutaneous Angiosarcoma
Angiosarcoma must be differentiated from other diseases that cause a highly vascular mass such as:[13]
- Atypical vascular lesions
- Hemangioma
- Glomangiosarcoma
- Carotid body tumor
- Malignant fibrous histiocytoma of soft tissue
Epidemiology and Demographics
Incidence
- In general 2% of soft tissue sarcomas are angiosarcomas, and the incidence of soft tissue sarcoma is about 6 per 100,000 person; on the other words,the incidence of angiosarcomas can be calculated approximately 1.2 per 1,000.000 person.[14][15]
Age
- Angiosarcoma is more commonly observed among patients aged between 40 to 75 years old.The peak age of incidence appears is the 7th decade,[16]
Gender
- Males are more commonly affected with angiosarcoma than females.[16]
- The male to female ratio is 2:1.[16]
Race
- There is no racial predilection for angiosarcoma. however, African-Americans in the U.S are rarely affected.[17]
Risk Factors
Common risk factors in the development of angiosarcoma include:[5]
- Chronic lymphedema
- Chronic exposure to polyvinyl chloride (PVC)
- Radiation exposure
- Exposure to Thorotrast
Natural History, Complications and Prognosis
Natural History
- The majority of patients with angiosarcoma remain asymptomatic for years.[5]
- Early clinical features may include nonspecific symptoms, such as pain, fatigue, malaise, and nausea.
- If left untreated, the majority of patients with angiosarcoma may rapidly progress to develop metastases.[16]
Complications
Common complications of angiosarcoma include:[5]
Prognosis
- Prognosis is generally poor; the 5-year survival rate of patients with angiosarcoma is approximately 12-33%.
- Poor prognostic factors include patient age (> 65 years), retroperitoneal location, and large tumor size.[16]
Diagnosis
Symptoms
- Angiosarcoma is usually asymptomatic and found incidentally.
- Symptoms of angiosarcoma are generally non-specific.
Physical Examination
Patients with angiosarcoma may appear cachectic or normal. In cutaneous angiosarcoma, physical examination findings may include:
- Bruise or skin ulceration
- Blushed purple-red papule
Laboratory Findings
There are no specific laboratory findings associated with angiosarcoma.
Imaging Findings
- The imaging modality of choice for angiosarcoma will depend on the location.
- For pulmonary angiosarcoma, the imaging modality of choice is enhanced CT scan.[16] For other types angiosarcoma, the imaging modality of choice is MRI.
CT
On CT, findings of angiosarcoma may include:[16]
- Vascular invasion
- Nodular enhancement (common)
- Hypoattenuating mass
- Multicentric lesions
-
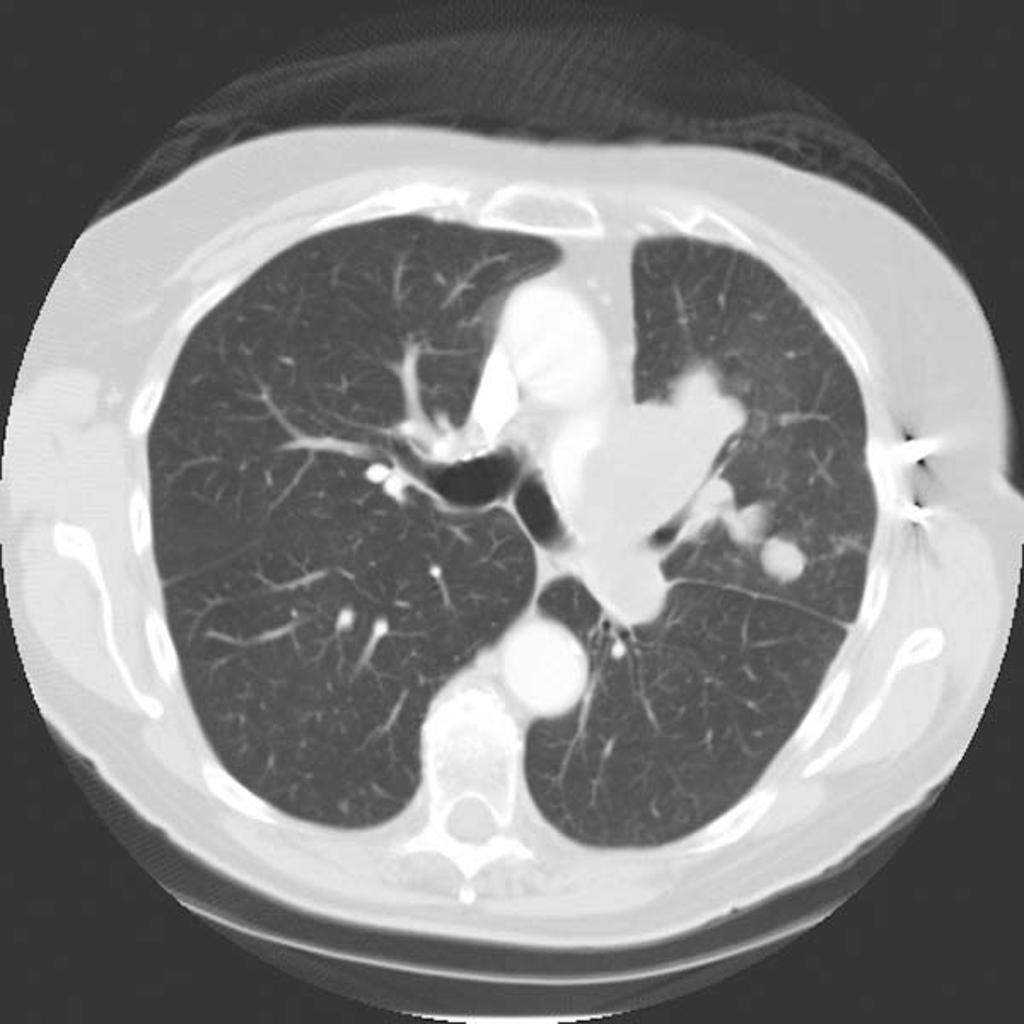
CT Pulmonary angiosarcoma
Courtesy of Radiopedia

MRI
On MRI, findings of angiosarcoma may include:
- T1/T2: heterogeneous areas of hyperintensity suggestive of a mixed tumour and hemorrhage
- T1 C+ (Gd): heterogeneous enhancement
Treatment
The mainstay of treatment for angiosarcoma is complete surgical resection with wide margins for local and locoregional disease in combination with preoperative or postoperative radiotherapy.[18] The role of adjuvant chemotherapy, is unclear. Adjuvant chemotherapy and/or radiotheray provide less mutilating surgery, and for patients with unresectable tumors or those who refuse surgery is an option.[19][20]
Medical Therapy
- The main adjuvant therapy for angiosarcoma is a doxorubicin-based regimen.[5] For angiosarcoma, doxorubicin monotherapy is indicated as first-line therapy.[5]
- Common complications of doxorubicin include:
- For patients with pulmonary angiosarcoma, a combination of radiotherapy and immunotherapy with recombinant interleukin-2 is the treatment of choice.[21] The response rate to chemotherapy in patients with angiosarcoma is poor.[5]
Surgery
Surgical resection in combination with radiation therapy is the treatment of choice for small angiosarcomas.[5]
- Surgical treatment for patients with cutaneous angiosarcoma is surgical resection with wide margins.[5] Surgery is not recommended on patients with large sized angiosarcomas. The recurrence rate of angiosarcoma after surgery is 80%.
Prevention
Primary Prevention
There are no primary preventive measures available for angiosarcoma.
Secondary Prevention
Once diagnosed and successfully treated, patients with angiosarcoma are followed-up every 3, 6, or 12 months depending on the stage at diagnosis. Follow-up testing for angiosarcoma may include:[5]
- Periodic imaging/angiography evaluation
- Laboratory testing: complete blood count (e.g., anemia)
References
- ↑ Barber W, Scriven P, Turner D, Hughes D, Wyld D (2010). "Epithelioid angiosarcoma: Use of angiographic embolisation and radiotherapy to control recurrent haemorrhage". J Surg Case Rep. 2010 (5): 7. doi:10.1093/jscr/2010.5.7. PMC 3649120. PMID 24946325.
- ↑ Matthew G. Fury, Cristina R. Antonescu, Kimberly J. Van Zee, Murray F. Brennan & Robert G. Maki (2005). "A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy". Cancer journal (Sudbury, Mass.). 11 (3): 241–247. PMID 16053668. Unknown parameter
|month=ignored (help) - ↑ M. Schlemmer, P. Reichardt, J. Verweij, J. T. Hartmann, I. Judson, A. Thyss, P. C. W. Hogendoorn, S. Marreaud, M. Van Glabbeke & J. Y. Blay (2008). "Paclitaxel in patients with advanced angiosarcomas of soft tissue: a retrospective study of the EORTC soft tissue and bone sarcoma group". European journal of cancer (Oxford, England : 1990). 44 (16): 2433–2436. doi:10.1016/j.ejca.2008.07.037. PMID 18771914. Unknown parameter
|month=ignored (help) - ↑ Marie Karanian & Jean-Michel Coindre (2015). "[Fourth edition of WHO classification tumours of soft tissue]". Annales de pathologie. 35 (1): 71–85. doi:10.1016/j.annpat.2014.11.003. PMID 25532684. Unknown parameter
|month=ignored (help) - ↑ 5.00 5.01 5.02 5.03 5.04 5.05 5.06 5.07 5.08 5.09 5.10 5.11 Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ (2010). "Angiosarcoma". Lancet Oncol. 11 (10): 983–91. doi:10.1016/S1470-2045(10)70023-1. PMID 20537949.
- ↑ Y. Amo, M. Masuzawa, Y. Hamada & K. Katsuoka (2004). "Serum concentrations of vascular endothelial growth factor-D in angiosarcoma patients". The British journal of dermatology. 150 (1): 160–161. PMID 14746640. Unknown parameter
|month=ignored (help) - ↑ Johanna Manner, Bernhard Radlwimmer, Peter Hohenberger, Katharina Mossinger, Stefan Kuffer, Christian Sauer, Djeda Belharazem, Andreas Zettl, Jean-Michel Coindre, Christian Hallermann, Jorg Thomas Hartmann, Detlef Katenkamp, Kathrin Katenkamp, Patrick Schoffski, Raf Sciot, Agnieszka Wozniak, Peter Lichter, Alexander Marx & Philipp Strobel (2010). "MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema". The American journal of pathology. 176 (1): 34–39. doi:10.2353/ajpath.2010.090637. PMID 20008140. Unknown parameter
|month=ignored (help) - ↑ Tianhua Guo, Lei Zhang, Ning-En Chang, Samuel Singer, Robert G. Maki & Cristina R. Antonescu (2011). "Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions". Genes, chromosomes & cancer. 50 (1): 25–33. doi:10.1002/gcc.20827. PMID 20949568. Unknown parameter
|month=ignored (help) - ↑ Anthony P. Fernandez, Yang Sun, Raymond R. Tubbs, John R. Goldblum & Steven D. Billings (2012). "FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations". Journal of cutaneous pathology. 39 (2): 234–242. doi:10.1111/j.1600-0560.2011.01843.x. PMID 22121953. Unknown parameter
|month=ignored (help) - ↑ S. A. Sinclair, L. Sviland & S. Natarajan (1998). "Angiosarcoma arising in a chronically lymphoedematous leg". The British journal of dermatology. 138 (4): 692–694. PMID 9640382. Unknown parameter
|month=ignored (help) - ↑ R. J. W. de Keizer, D. de Wolff-Rouendaal & M. A. Nooy (2008). "Angiosarcoma of the eyelid and periorbital region. Experience in Leiden with iridium192 brachytherapy and low-dose doxorubicin chemotherapy". Orbit (Amsterdam, Netherlands). 27 (1): 5–12. doi:10.1080/01676830601168926. PMID 18307140.
- ↑ Rita Vora, Gopikrishnan Anjaneyan & Rajat Gupta (2014). "Cutaneous angiosarcoma of head and neck". Indian journal of dermatology. 59 (6): 632. doi:10.4103/0019-5154.143575. PMID 25484419. Unknown parameter
|month=ignored (help) - ↑ C. D. Fletcher, A. Beham, S. Bekir, A. M. Clarke & N. J. Marley (1991). "Epithelioid angiosarcoma of deep soft tissue: a distinctive tumor readily mistaken for an epithelial neoplasm". The American journal of surgical pathology. 15 (10): 915–924. PMID 1718176. Unknown parameter
|month=ignored (help) - ↑ Robin J. Young, Nicola J. Brown, Malcolm W. Reed, David Hughes & Penella J. Woll (2010). "Angiosarcoma". The Lancet. Oncology. 11 (10): 983–991. doi:10.1016/S1470-2045(10)70023-1. PMID 20537949. Unknown parameter
|month=ignored (help) - ↑ Andrea Ferrari, Iyad Sultan, Tseng Tien Huang, Carlos Rodriguez-Galindo, Ahmad Shehadeh, Cristina Meazza, Kirsten K. Ness, Michela Casanova & Sheri L. Spunt (2011). "Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database". Pediatric blood & cancer. 57 (6): 943–949. doi:10.1002/pbc.23252. PMID 21793180. Unknown parameter
|month=ignored (help) - ↑ 16.0 16.1 16.2 16.3 16.4 16.5 16.6 Sturgis EM, Potter BO. Sarcomas of the head and neck region. Curr Opin Oncol. 2003 May. 15(3):239-52
- ↑ Erich M. Sturgis & Bryan O. Potter (2003). "Sarcomas of the head and neck region". Current opinion in oncology. 15 (3): 239–252. PMID 12778019. Unknown parameter
|month=ignored (help) - ↑ W. M. Lydiatt, A. R. Shaha & J. P. Shah (1994). "Angiosarcoma of the head and neck". American journal of surgery. 168 (5): 451–454. PMID 7977971. Unknown parameter
|month=ignored (help) - ↑ Robin J. Young, Nicola J. Brown, Malcolm W. Reed, David Hughes & Penella J. Woll (2010). "Angiosarcoma". The Lancet. Oncology. 11 (10): 983–991. doi:10.1016/S1470-2045(10)70023-1. PMID 20537949. Unknown parameter
|month=ignored (help) - ↑ Guy Lahat, Asha R. Dhuka, Hen Hallevi, Lianchun Xiao, Changye Zou, Kerrington D. Smith, Thuy L. Phung, Raphael E. Pollock, Robert Benjamin, Kelly K. Hunt, Alexander J. Lazar & Dina Lev (2010). "Angiosarcoma: clinical and molecular insights". Annals of surgery. 251 (6): 1098–1106. doi:10.1097/SLA.0b013e3181dbb75a. PMID 20485141. Unknown parameter
|month=ignored (help) - ↑ Duck L, Baurain JF, Machiels JP (2004). "Treatment of a primary pulmonary angiosarcoma". Chest. 126 (1): 317–8, author reply 318. doi:10.1378/chest.126.1.317. PMID 15249484.