Hemochromatosis pathophysiology
| https://https://www.youtube.com/watch?v=iUga3oM65Bc%7C350}} |
|
Hemochromatosis Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Hemochromatosis pathophysiology On the Web |
|
American Roentgen Ray Society Images of Hemochromatosis pathophysiology |
|
Risk calculators and risk factors for Hemochromatosis pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief:
Overview
Pathophysiology

In the normal liver, iron is present at a concentration lower than 20 μmol/g of dry weight. Iron is normally lost in sweat, shed skin cells, and the gastrointestinal tract at a rate of approximately 1 mg/day. Premenopausal adult women lose an additional 0.5 to 1.0 mg/day because of menses. These losses are usually balanced by the absorption of 10 percent of the 10 to 20 mg of iron in the diet in Western societies. For example, HFE is only part of the story, since many patients with mutated HFE do not manifest clinical iron overload, and some patients with iron overload have a normal HFE genotype.
In general, people with abnormal iron regulatory genes do not reduce their absorption of iron in response to increased iron levels in the body. Thus the iron stores of the body increase. As they increase the iron which is initially stored as ferritin is deposited in organs as haemosiderin and this is toxic to tissue, probably at least partially by inducing oxidative stress.[1].
Iron is a pro-oxidant. Thus, hemochromatosis shares common symptomology (e.g., cirrhosis and dyskinetic symptoms) with other "pro-oxidant" diseases such as Wilson's disease, chronic manganese poisoning, and hyperuricemic syndrome in Dalmatian dogs. The latter also experience "bronzing".
Intestinal crypt enterocytes and iron overload
The sensor pathway inside the small bowel enterocyte can be disrupted due to genetic errors in the iron regulatory apparatus. The enterocyte in the small bowel crypt must somehow sense the amount of circulating iron. Depending on this information, the enterocyte cell can regulate the quantity of iron receptors and channel proteins. If there is little iron, the enterocyte cell will express many of these proteins. If there is a lot, the cell will turn off the expression of iron transporters.
In haemochromatosis, the enterocyte is somehow constantly fooled into thinking there is iron depletion. As a consequence, it overexpresses the necessary channel proteins, this leading to a massive, and unnecessary iron absorption. Details of how this process exactly works in health and disease are still being discovered as of May, 2007.
These iron transport proteins are named DMT-1 (divalent metal transporter), for the luminal side of the cell, and ferroportin, the only known cellular iron exporter, for the basal side of the cell.
Hepcidin-ferroportin axis and iron overload
Recently, a new unifying theory for the pathogenesis of hereditary hemochromatosis has been proposed that focuses on the hepcidin-ferroportin regulatory axis. Inappropriately low levels of hepcidin, the iron regulatory hormone, can account for the clinical phenotype of iron overload. In this theory, low levels of circulating hepcidin result in higher levels of ferroportin expression in intestinal enterocytes and reticuloendothelial macrophages. As a result, this causes iron accumulation. HFE, hemojuvelin, BMP's and TFR2 are implicated in regulating hepcidin expression.
Genetics
The regulation of how much iron enters the body from food is complex, and each year brings new discoveries about the numerous factors working in harmony to bring about balance in the metabolism of iron in humans
One of the best-characterized genes that regulates the amount of iron absorbed from food is called HFE. The HFE gene has two common mutations, C282Y and H63D.
Inheriting just one of the C282Y mutations (heterozygous) makes a person a carrier who can pass this mutation onward. Carriers of one HFE mutation ordinarily do not manifest with clinically relevant iron accumulation at all.
-

Haemochromatosis types 1-3 are inherited in an autosomal recessive fashion.
-
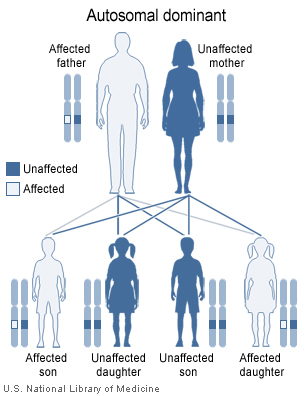
Haemochromatosis type 4 is inherited in an autosomal dominant fashion.


In the United States, most people with clinically measureable haemochromatosis (i.e., iron overload with or without end organ damage) have inherited two copies of C282Y — one from each parent — and are therefore homozygous for the trait. Mutations of the HFE gene account for 90% of the cases of clinical iron overload. This gene is closely linked to the HLA-A3 locus. Homozygosity for the C282Y mutation is the most prevalent condition resulting in clinical iron accumulation, although heterozygosity for C282Y/H63D mutations, so-called compound heterozygotes, is also known to cause clinical iron overload. So, both homozygotes for C282Y and compound heterozygotes for C282Y/H63D are known to have clinical iron overload and hemochromatosis.
Most people with two copies of C282Y or one copy each of C282Y/H63D do not manifest clinical hemochromatosis, a phenomenon known as low incomplete penetration. [2] Penetration differs between different populations.
Other genes whose mutations have been associated with iron overload include the autosomal dominant SLC11A3/ferroportin 1 gene and TfR2 (transferrin receptor 2). These mutations, and the iron overload they cause, are much rarer than HFE-haemochromatosis.
Microscopic Pathology
Liver biopsy - Liver biopsies involve taking a sample of tissue from the liver, using a thin needle. The amount of iron in the sample is then quantified and compared to normal, and evidence of liver damage, especially cirrhosis, measured microscopically. Formerly, this was the only way to confirm a diagnosis of hemochromatosis but measures of transferrin and ferritin along with a history are considered adequate in determining the presence of the malady. Risks of biopsy include bruising, bleeding and infection. Now, when a history and measures of transferrin or ferritin point to haemochromatosis, it is debatable whether a liver biopsy is still necessary to quantify the amount of accumulated iron.
References
- ↑ Shizukuda Y, Bolan C, Nguyen T, Botello G, Tripodi D, Yau Y, Waclawiw M, Leitman S, Rosing D (2007). "Oxidative stress in asymptomatic subjects with hereditary hemochromatosis". Am J Hematol. 82 (3): 249–50. PMID 16955456.
- ↑ Olynyk J, Cullen D, Aquilia S, Rossi E, Summerville L, Powell L (1999). "A population-based study of the clinical expression of the hemochromatosis gene". N Engl J Med. 341 (10): 718–24. PMID 10471457.