Familial hyperchylomicronemia
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]Associate Editor(s)-in-Chief: Vishal Devarkonda, M.B.B.S[2]
Synonyms and keywords: Type I hyperlipoproteinemia, Burger-Grutz syndrome, Primary hyperlipoproteinemia, lipoprotein lipase deficiency, LPL deficiency, Idiopathic hyperlipemia, Essential hyperlipemia, Familial hyperlipemia, Lipase D deficiency, Hyperlipoproteinemia type IA, Familial chylomicronemia, Familial lipoprotein lipase deficiency, and Familial hyperchylomicronemia.
Overview
This very rare form is due to a deficiency of lipoprotein lipase (LPL) or altered apolipoprotein C2, resulting in elevated chylomicron which are the particles that transfer fatty acids from the digestive tract to the liver. Lipoprotein lipase is also responsible for the initial breakdown of endogenously made triacylglycerides in the form of very low density lipoprotein (VLDL). As such, one would expect a defect in LPL to also result in elevated VLDL. Its prevalence is one in 1,000,000 population.
Classification
- Type I hyperlipoproteinemia can be further classified into:
Type 1A
It occurs due to deficiency of lipoprotein lipase enzyme.
Type 1B
Altered apolipoprotein C2 causes type 1B hyperlipoproteinemia.
Type 1C
Presence of LPL inhibitor is the cause of type 1C hyperlipoproteinemia.
Historical Perspective
- In 1932, Familial LPL deficency was first described by Burger and Grutz[1].
- In 1967, Fredrickson classified and described hyperlipidemias, using paper electrophoresis[2].
Pathophysiology
- Type I hyperlipoproteinemia is a rare autosomal recessive disorder of lipoprotein metabolism[3][4][5].
Pathogenesis
- Lipoprotein lipase(LPL) hydrolysis Triglyceride-rich lipoproteins (TG) such as chylomicrons and very low-density lipoproteins. It catalyzes, the removal of TG from bloodstream generating free fatty acids for tissues.
- For full enzymatic activity, LPL requires following cofactors:-
- Apolipoprotein C-II and apolipoprotein A-V that are LPL activators
- Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein
- Lipase maturation factor 1
- Development of Type I hyperlipoproteinemia is the result of the functional mutations in one of all these genes results in type I hyperlipoproteinemia.
Type 1C (Familial lipoprotein lipase inhibitor)
- Familial lipoprotein lipase inhibitor seems to be inherited as an autosomal dominant trait[6].
- Presence of familial lipase inhibitor, inhibit the action of lipoprotein lipase, resulting in decreased postheparin plasma LPL activity, elevated adipose tissue LPL activity, and normal plasma levels of functional apoC-I1.
- Functionally inactive or absent lipoprotein lipase enzyme, results in massive accumulation of chylomicrons, with extremely high level of plasma triglycerides.
Causes
The cause of type 1 hyperlipidemia remains genetic[7].
Differential diagnosis
| Diseases | Laboratory Findings | Physical Examination | History and symptoms | other findings |
|---|---|---|---|---|
| Familial combined hyperlipidemia | ||||
| Monogenic familial hypertriglyceridemia | ||||
| Secondary causes of hypertriglyceridemia | ||||
| Diabetes mellitus | ||||
| Paraproteinemic disorders | ||||
| Alcohol usage | ||||
| Estrogen thearapy | ||||
| Glucocorticoids | ||||
| Isotretinoin | ||||
| Antihypertensive agents |
Epidemiology and Demographics
Epidemiology
- The disease has been described in all races.
- The prevalence is much higher in some areas of Quebec, Canada, as a result of a founder effect.
- The prevalence of familial LPL deficiency is approximately one in 1,000,000 in the general US population.
Demographics
Age
- 25% of affected children develop symptoms before one year of age.
- Majority develop symptoms before ten years of age.
- Few individuals develop symptoms, at the time of pregnancy.
Gender
- Males and females are equally affected.
Screening
- There are no screening guidelines for Familial hyperchylomicronemia[7][3][8].
- Evaluation of Relatives at Risk.It is appropriate to measure plasma triglyceride concentration in at-risk sibs during infancy; early diagnosis and implementation of dietary fat intake restriction can prevent symptoms and related medical complications.
Natural History, Complications, and Prognosis
Natural History
- If left untreated, pancreatitis can develop into a chronic condition that can damage the pancreas and, in rare cases, be life-threatening[8].
Complications
- Pancreatitis and recurrent episodes of abdominal pain may develop[8].
- Xanthomas are not usually painful unless they are rubbed a lot.
Prognosis
- People with this condition who follow a very low-fat diet can live into adulthood[8].
Diagnosis
- Presumptive diagnosis can be made, when an infant presents with a history of failure to thrive or recurrent abdominal pain, with an documented high fasting plasma triglyceride concentration[9][8].
- Diagnosis of familial lipoprotein lipase deficiency is confirmed by detection of low or absent LPL enzyme activity in an assay system that contains either normal plasma or apoprotein C-II excluding hepatic lipase.
History and symptoms
Symptoms of Type I hyperlipoproteinemia include[7][10][8]:-
- Abdominal pain (may appear as colic in infancy)
- Loss of appetite
- Nausea
- Pain in the muscles and bones (musculoskeletal pain)
- Vomiting
- Small yellow papules localized over the trunk, buttocks, knees, and extensor surfaces of the arms
- In rare cases, neurological features develop, including depression, memory loss, and mild intellectual decline (dementia).
Physical examination
- Signs of Type 1 hyperlipoproteinemia include[7][10][8]:-
- Enlarged liver and spleen
- Failure to thrive in infancy
- Fatty deposits in the skin (xanthomas)
- Pale retinas and white-colored blood vessels in the retinas
- Pancreatitis that keeps returning
- Yellowing of the eyes and skin (jaundice)
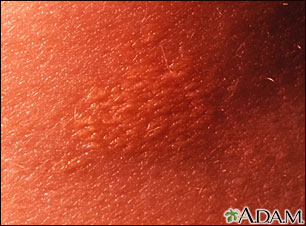
Xanthoma - close-up

Laboratory finding
- Laboratory findings consistent with the Type I hyperlipoproteinemia include the following:-
| Laboratory finding | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phenotype | Lipoprotein(s)
Elevated |
Serum total
cholesterol |
HDL | VLDL | Serum
triglycerides |
Plasma
appearance |
Postheparin
lipolytic
|
Glucose
tolerance |
Carbohydrate
inducibility |
Fat tolerance |
| Hyperlipoproteinemia type 1 | Chylomicrons(↑↑↑↑) | Normal to
elevated |
↓↓↓ | ↓ | ↑↑↑↑ | Creamy | Decreased | Normal | May be abnormal | Markedly abnormal |
Molecular Genetic Testing
- Diagnosis can be confirmed by molecular genetic testing that can detect mutations in the LPL gene.[3]
- The test is often not necessary to confirm a diagnosis of LPLD.
Treatment
- Treatment for hyperlipoproteinemia type 1 is intended to control blood triglyceride levels with a very low-fat diet[7][9] [8].
- It is recommended that individuals with this condition eat no more than 20 grams of fat per day.
- Medium-chain fatty acids (such as coconut oil) can be incorporated into the diet, as they are absorbed by the body in a different manner.
- Dietary counseling may be helpful to maintain adequate calorie and nutrient intake.
Pregnancy Management
- Pregnant women may experience significant changes in lipid level in second and third trimester, and may require strategies to lower fat intake.
- Periodic assessment of plasma triglycerides is highly recommended[11] .
- Comprehensive analysis of risks versus benefits is required before the use of fibrates, nicotinic acid and omega-3 fatty acid[11].
Investigative Therapies
- Orlistat in conjunction with a low fat diet has been used to treat some patients with compound heterozygous LPLD[12].
Gene Therapy
- Alipogene tipavovec(Glybera) gene therapy was approved by European commission(2012), in treating adult patients with recurrent episodes of pancreatitis[13].
Prevention
- There is no way to prevent someone from inheriting this syndrome[7].
- Genetic counseling is recommended for patients and family members as well.
Prevention of Primary Manifestations
- Maintaining the plasma triglyceride concentration at less than 2000 mg/dl keeps the individual with familial LPL deficiency free of symptoms. This can be accomplished by restriction of dietary fat to no more than 20 g/day or 15% of total energy intake[8].
- Periodic assessment of plasma triglycerides levels is highly recommended.
- Patients should avoid agents that increased endogenous triglyceride levels like alcohol, diuretics, oral estrogens, isoretinoin, glucorticords, and beta-blockers.
Prevention of Secondary Complications
- Prevention of acute recurrent pancreatitis decreases the risk of development of diabetes mellitus [8] and fat malabsorption .
References
- ↑ Template:Http://rarediseases.org/rare-diseases/familial-lipoprotein-lipase-deficiency/Accessed on 7 November,2016
- ↑ Culliton BJ (1987). "Fredrickson's bitter end at Hughes". Science. 236 (4807): 1417–8. PMID 3296193.
- ↑ 3.0 3.1 3.2 3.3 Pingitore P, Lepore SM, Pirazzi C, Mancina RM, Motta BM, Valenti L; et al. (2016). "Identification and characterization of two novel mutations in the LPL gene causing type I hyperlipoproteinemia". J Clin Lipidol. 10 (4): 816–23. doi:10.1016/j.jacl.2016.02.015. PMID 27578112.
- ↑ Young SG, Zechner R (2013). "Biochemistry and pathophysiology of intravascular and intracellular lipolysis". Genes Dev. 27 (5): 459–84. doi:10.1101/gad.209296.112. PMC 3605461. PMID 23475957.
- ↑ Pasalić D, Jurcić Z, Stipancić G, Ferencak G, Leren TP, Djurovic S; et al. (2004). "Missense mutation W86R in exon 3 of the lipoprotein lipase gene in a boy with chylomicronemia". Clin Chim Acta. 343 (1–2): 179–84. doi:10.1016/j.cccn.2004.01.029. PMID 15115692.
- ↑ Francis A, Levy Y (2002). "[Chylomicronemia syndrome]". Harefuah. 141 (2): 201–3, 221, 220. PMID 11905095.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 https://rarediseases.info.nih.gov/diseases/6414/hyperlipoproteinemia-type-1 Accessed on 7 November,2016
- ↑ 8.0 8.1 8.2 8.3 8.4 8.5 8.6 8.7 8.8 8.9 Template:Https://medlineplus.gov/ency/article/000408htm Accessed on 7 November,2016
- ↑ 9.0 9.1 Template:Https://www.ncbi.nlm.nih.gov/books/NBK1308/ Accessed on 7 November,2016
- ↑ 10.0 10.1 Robinson JG (2012). "What is the role of advanced lipoprotein analysis in practice?". J Am Coll Cardiol. 60 (25): 2607–15. doi:10.1016/j.jacc.2012.04.067. PMID 23257303.
- ↑ 11.0 11.1 Al-Shali K, Wang J, Fellows F, Huff MW, Wolfe BM, Hegele RA (2002). "Successful pregnancy outcome in a patient with severe chylomicronemia due to compound heterozygosity for mutant lipoprotein lipase". Clin Biochem. 35 (2): 125–30. PMID 11983347.
- ↑ Blackett P, Tryggestad J, Krishnan S, Li S, Xu W, Alaupovic P; et al. (2013). "Lipoprotein abnormalities in compound heterozygous lipoprotein lipase deficiency after treatment with a low-fat diet and orlistat". J Clin Lipidol. 7 (2): 132–9. doi:10.1016/j.jacl.2012.11.006. PMID 23415432.
- ↑ Gaudet D, Stroes ES, Méthot J, Brisson D, Tremblay K, Bernelot Moens SJ; et al. (2016). "Long-Term Retrospective Analysis of Gene Therapy with Alipogene Tiparvovec and Its Effect on Lipoprotein Lipase Deficiency-Induced Pancreatitis". Hum Gene Ther. doi:10.1089/hum.2015.158. PMID 27412455.