Ibrutinib
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Aparna Vuppala, M.B.B.S. [2]
Disclaimer
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Ibrutinib is an kinase inhibitor that is FDA approved for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. Common adverse reactions include thrombocytopenia, diarrhea, neutropenia, anemia, fatigue, musculoskeletal pain, peripheral edema, upper respiratory tract infection, nausea, bruising, dyspnea, constipation, rash, abdominal pain, vomiting and decreased appetite ..
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Mantle Cell Lymphoma
- Ibrutinib is indicated for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
- Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
Chronic Lymphocytic Leukemia
- Ibrutinib is indicated for the treatment of patients with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy .
Chronic Lymphocytic Leukemia with 17p deletion
- Ibrutinib is indicated for the treatment of patients with chronic lymphocytic leukemia (CLL) with 17p deletion.
Waldenström's Macroglobulinemia
- Ibrutinib is indicated for the treatment of patients with Waldenström's macroglobulinemia (WM)
Dosing Guidelines
- Administer Ibrutinib orally once daily at approximately the same time each day. Swallow the capsules whole with water. Do not open, break, or chew the capsules.
Dosage
Mantle Cell Lymphoma
- The recommended dose of Ibrutinib for MCL is 560 mg (four 140 mg capsules) orally once daily.
- The recommended dose of Ibrutinib for CLL and WM is 420 mg (three 140 mg capsules) orally once daily.
Dose Modifications for Adverse Reactions
- Interrupt Ibrutinib therapy for any Grade 3 or greater non-hematological, Grade 3 or greater neutropenia with infection or fever, or Grade 4 hematological toxicities. Once the symptoms of the toxicity have resolved to Grade 1 or baseline (recovery), Ibrutinib therapy may be reinitiated at the starting dose. If the toxicity reoccurs, reduce dose by one capsule (140 mg per day). A second reduction of dose by 140 mg may be considered as needed. If these toxicities persist or recur following two dose reductions, discontinue Ibrutinib.
- Recommended dose modifications are described below:
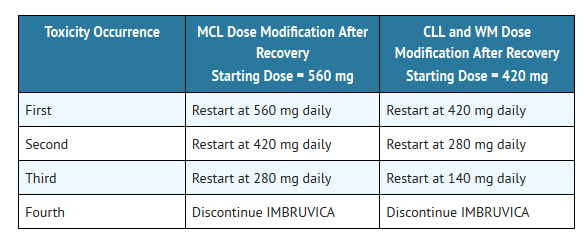
This image is provided by the National Library of Medicine.

- Dose Modifications for Use with CYP3A Inhibitors
- Avoid co-administration with strong or moderate CYP3A inhibitors and consider alternative agents with less CYP3A inhibition.
- Concomitant use of strong CYP3A inhibitors which would be taken chronically (e.g., ritonavir, indinavir, nelfinavir, saquinavir, boceprevir, telaprevir, nefazodone) is not recommended. For short-term use (treatment for 7 days or less) of strong CYP3A inhibitors (e.g., antifungals and antibiotics) consider interrupting Ibrutinib therapy until the CYP3A inhibitor is no longer needed.
- Reduce Ibrutinib dose to 140 mg if a moderate CYP3A inhibitor must be used (e.g., fluconazole, darunavir, erythromycin, diltiazem, atazanavir, aprepitant, amprenavir, fosamprevir, crizotinib, imatinib, verapamil, and ciprofloxacin).
- Patients taking concomitant strong or moderate CYP3A inhibitors should be monitored more closely for signs of Ibrutinib toxicity.
Dose Modifications for Use in Hepatic Impairment
- For patients with mild liver impairment (Child-Pugh class A), the recommended dose is 140 mg daily (one capsule). Avoid the use of Ibrutinib in patients with moderate or severe hepatic impairment (Child-Pugh classes B and C)
Missed Dose
- If a dose of Ibrutinib is not taken at the scheduled time, it can be taken as soon as possible on the same day with a return to the normal schedule the following day. Extra capsules of Ibrutinib should not be taken to make up for the missed dose.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Ibrutinib in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Ibrutinib in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Ibrutinib in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Ibrutinib in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Ibrutinib in pediatric patients.
Contraindications
- None
Warnings
Hemorrhage
- Fatal bleeding events have occurred in patients treated with Ibrutinib. Grade 3 or higher bleeding events (subdural hematoma, gastrointestinal bleeding, hematuria and post procedural hemorrhage) have occurred in up to 6% of patients. Bleeding events of any grade, including bruising and petechiae, occurred in approximately half of patients treated with Ibrutinib.
- The mechanism for the bleeding events is not well understood.
- Ibrutinib may increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies.
- Consider the benefit-risk of withholding Ibrutinib for at least 3 to 7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding .
Infections
- Fatal and non-fatal infections have occurred with Ibrutinib therapy. Grade 3 or greater infections occurred in 14% to 26% of patients. Cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients treated with Ibrutinib. Monitor patients for fever and infections and evaluate promptly.
Cytopenias
- Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (range, 19 to 29%), thrombocytopenia (range, 5 to 17%), and anemia (range, 0 to 9%) occurred in patients treated with Ibrutinib.
- Monitor complete blood counts monthly.
Atrial Fibrillation
- Atrial fibrillation and atrial flutter (range, 6 to 9%) have occurred in patients treated with Ibrutinib, particularly in patients with cardiac risk factors, acute infections, and a previous history of atrial fibrillation. Periodically monitor patients clinically for atrial fibrillation. Patients who develop arrhythmic symptoms (e.g., palpitations, lightheadedness) or new onset dyspnea should have an ECG performed. If atrial fibrillation persists, consider the risks and benefits of Ibrutinib treatment and dose modification .
Second Primary Malignancies
- Other malignancies (range, 5 to 14%) including non-skin carcinomas (range, 1 to 3%) have occurred in patients treated with Ibrutinib. The most frequent second primary malignancy was non-melanoma skin cancer (range, 4 to 11 %).
Tumor Lysis Syndrome
- Tumor lysis syndrome has been reported with Ibrutinib therapy. Monitor patients closely and take appropriate precautions in patients at risk for tumor lysis syndrome (e.g. high tumor burden).
Embryo-Fetal Toxicity
- Based on findings in animals, Ibrutinib can cause fetal harm when administered to a pregnant woman. Ibrutinib caused malformations in rats at exposures 14 times those reported in patients with MCL and 20 times those reported in patients with CLL or WM, receiving the ibrutinib dose of 560 mg per day and 420 mg per day, respectively. Reduced fetal weights were observed at lower exposures. Advise women to avoid becoming pregnant while taking Ibrutinib. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus
Adverse Reactions
Clinical Trials Experience
- The following adverse reactions are discussed in more detail in other sections of the labeling:
- Hemorrhage
- Infections
- Cytopenias
- Atrial Fibrillation
- Second Primary Malignancies
- Tumor Lysis Syndrome
- Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Experience
Mantle Cell Lymphoma
- The data described below reflect exposure to Ibrutinib in a clinical trial that included 111 patients with previously treated MCL treated with 560 mg daily with a median treatment duration of 8.3 months.
- The most commonly occurring adverse reactions (≥ 20%) were thrombocytopenia, diarrhea, neutropenia, anemia, fatigue, musculoskeletal pain, peripheral edema, upper respiratory tract infection, nausea, bruising, dyspnea, constipation, rash, abdominal pain, vomiting and decreased appetite (seeTABLES 1 and 2).
- The most common Grade 3 or 4 non-hematological adverse reactions (≥ 5%) were pneumonia, abdominal pain, atrial fibrillation, diarrhea, fatigue, and skin infections.
- Fatal and serious cases of renal failure have occurred with Ibrutinib therapy. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 9% of patients.
- Adverse reactions from the MCL trial (N=111) using single agent Ibrutinib 560 mg daily occurring at a rate of ≥ 10% are presented in Table 1.
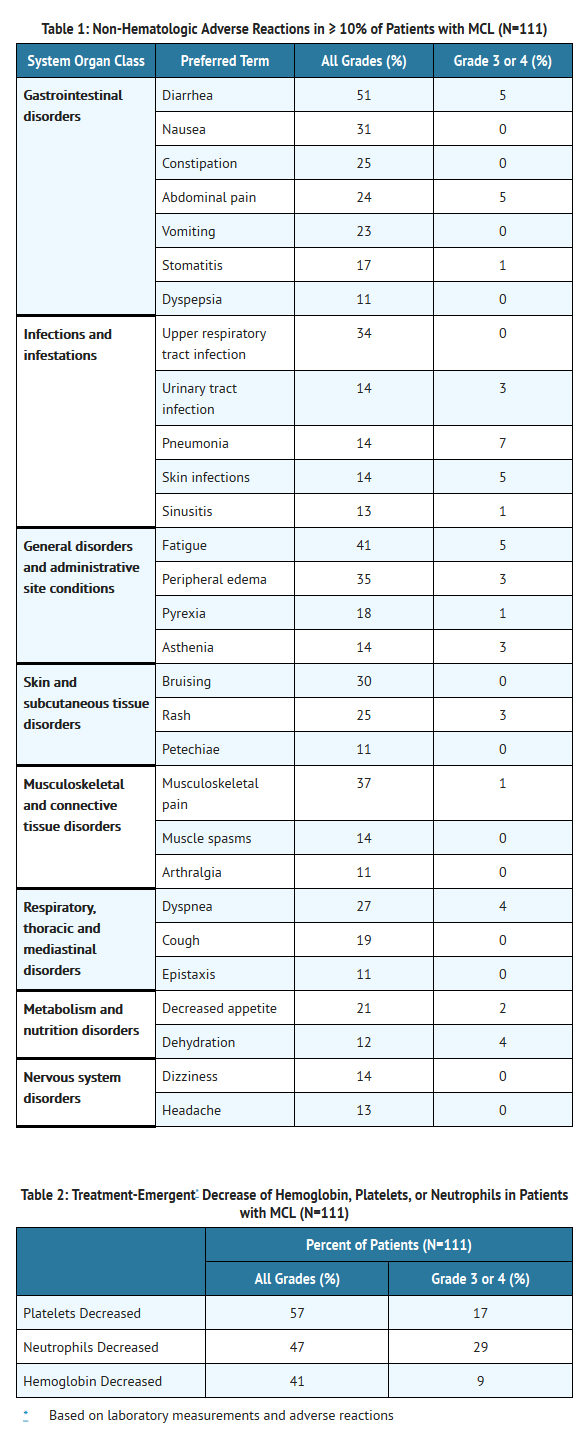
This image is provided by the National Library of Medicine.

- Ten patients (9%) discontinued treatment due to adverse reactions in the trial (N=111). The most frequent adverse reaction leading to treatment discontinuation was subdural hematoma (1.8%). Adverse reactions leading to dose reduction occurred in 14% of patients.
- Patients with MCL who develop lymphocytosis greater than 400,000/mcL have developed intracranial hemorrhage, lethargy, gait instability, and headache. However, some of these cases were in the setting of disease progression.
- Forty percent of patients had elevated uric acid levels on study including 13% with values above 10 mg/dL. Adverse reaction of hyperuricemia was reported for 15% of patients.
Chronic Lymphocytic Leukemia
- The data described below reflect exposure to Ibrutinib in an open label clinical trial (Study 1) that included 48 patients with previously treated CLL and a randomized clinical trial (Study 2) that included 391 randomized patients with previously treated CLL or SLL.
- The most commonly occurring adverse reactions in Study 1 and Study 2 (≥ 20%) were thrombocytopenia, neutropenia, diarrhea, anemia, fatigue, musculoskeletal pain, upper respiratory tract infection, rash, nausea, and pyrexia.
- Approximately five percent of patients receiving Ibrutinib in Study 1 and Study 2 discontinued treatment due to adverse events. These included infections, subdural hematomas and diarrhea. Adverse events leading to dose reduction occurred in approximately 6% of patients.
Study 1
- Adverse reactions and laboratory abnormalities from the CLL trial (N=48) using single agent Ibrutinib 420 mg daily occurring at a rate of ≥ 10% are presented in Tables 3 and 4.
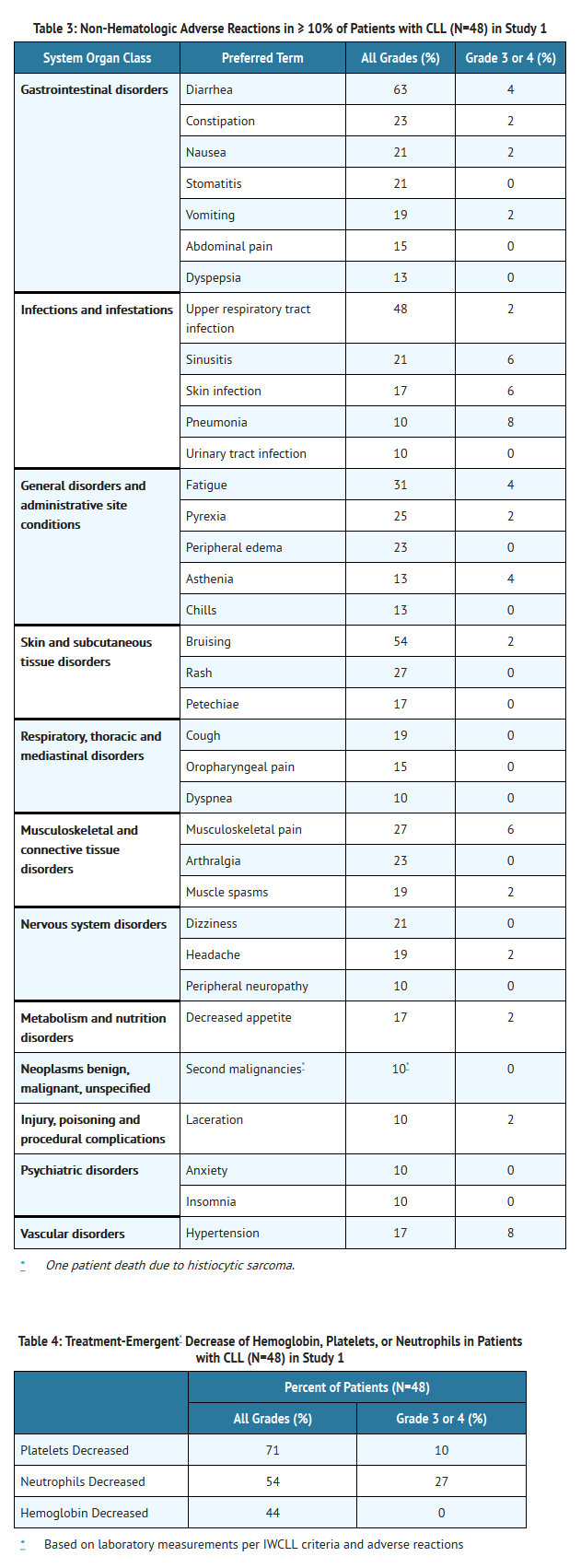
This image is provided by the National Library of Medicine.

Study 2
- Adverse reactions and laboratory abnormalities described below in Tables 5 and 6 reflect exposure to Ibrutinib with a median duration of 8.6 months and exposure to ofatumumab with a median of 5.3 months in Study 2.
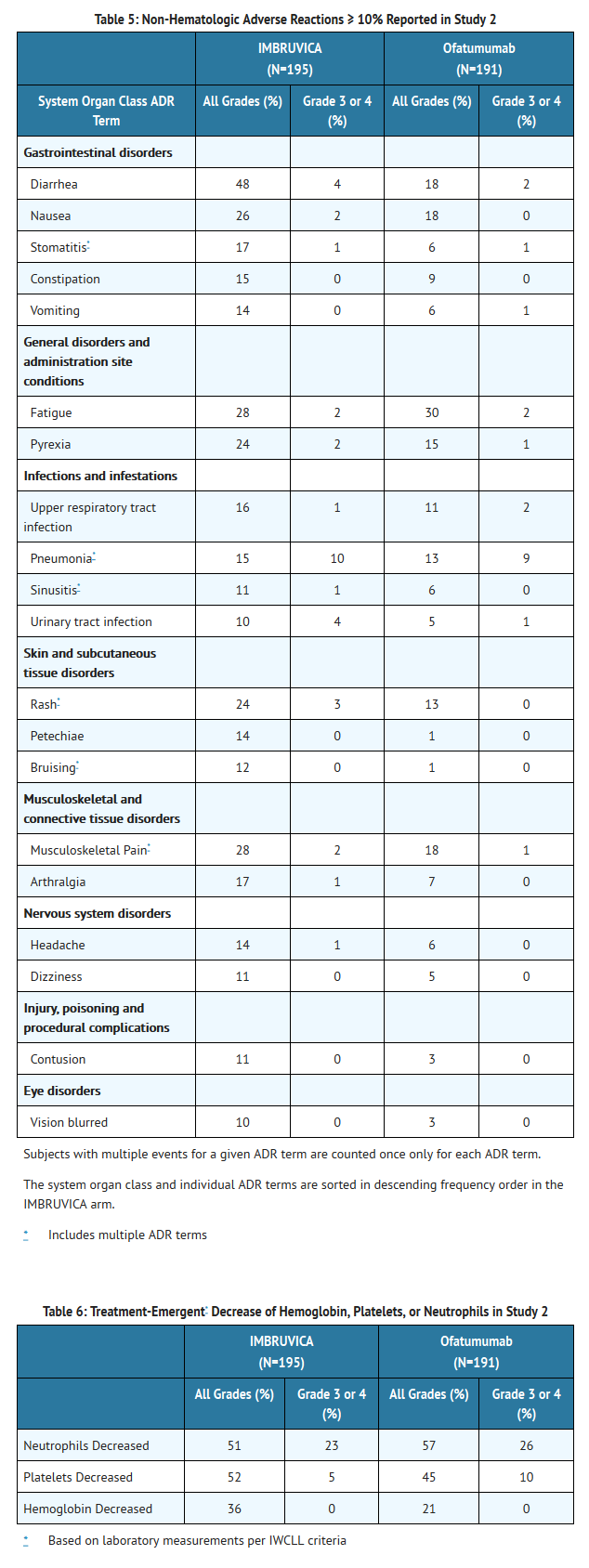
This image is provided by the National Library of Medicine.

Waldenström's Macroglobulinemia
- The data described below reflect exposure to Ibrutinib in an open label clinical trial that included 63 patients with previously treated WM.
- The most commonly occurring adverse reactions in the WM trial (≥ 20%) were neutropenia, thrombocytopenia, diarrhea, rash, nausea, muscle spasms, and fatigue.
- Six percent of patients receiving Ibrutinib in the WM trial discontinued treatment due to adverse events. Adverse events leading to dose reduction occurred in 11% of patients.
- Adverse reactions and laboratory abnormalities described below in Tables 7 and 8 reflect exposure to Ibrutinib with a median duration of 11.7 months in the WM trial.
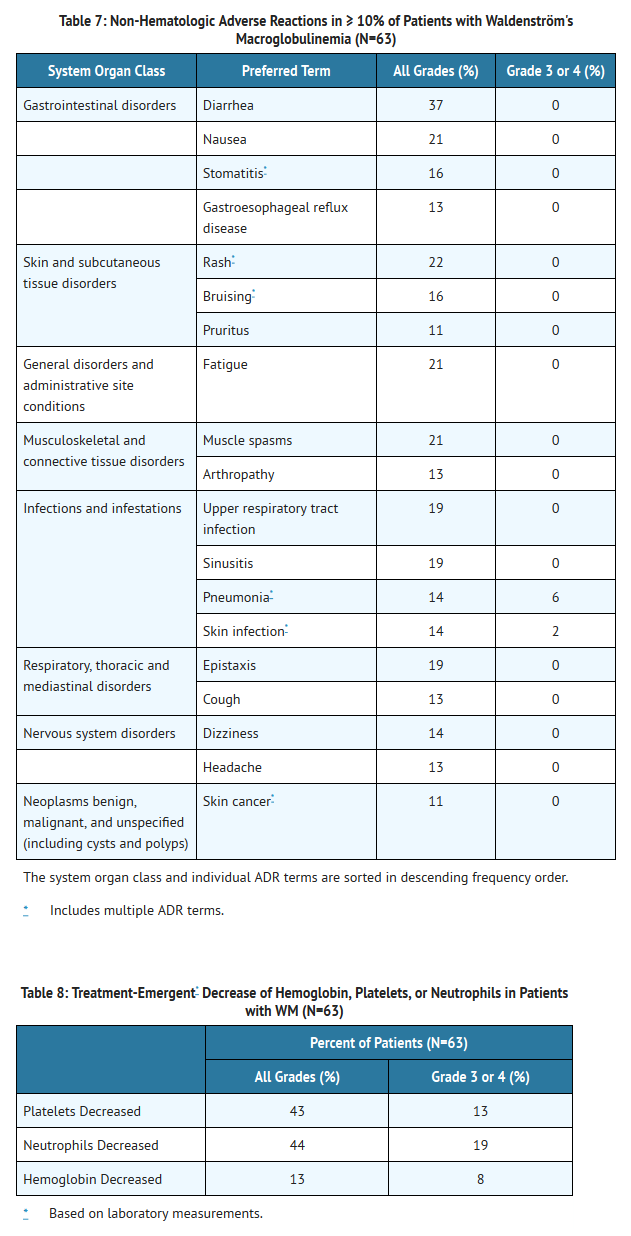
This image is provided by the National Library of Medicine.

Postmarketing Experience
- The following adverse reactions have been identified during post-approval use of Ibrutinib. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity reactions including anaphylactic shock (fatal), urticaria, and angioedema have been reported.
Drug Interactions
- Ibrutinib is primarily metabolized by cytochrome P450 enzyme 3A.
CYP3A Inhibitors
- In healthy volunteers, co-administration of ketoconazole, a strong CYP3A inhibitor, increased Cmax and AUC of ibrutinib by 29- and 24-fold, respectively. The highest ibrutinib dose evaluated in clinical trials was 12.5 mg/kg (actual doses of 840 – 1400 mg) given for 28 days with single dose AUC values of 1445 ± 869 ng ∙ hr/mL which is approximately 50% greater than steady state exposures seen at the highest indicated dose (560 mg).
- Avoid concomitant administration of Ibrutinib with strong or moderate inhibitors of CYP3A. For strong CYP3A inhibitors used short-term (e.g., antifungals and antibiotics for 7 days or less, e.g., ketoconazole, itraconazole, voriconazole, posaconazole, clarithromycin, telithromycin) consider interrupting Ibrutinib therapy during the duration of inhibitor use. Avoid strong CYP3A inhibitors that are needed chronically. If a moderate CYP3A inhibitor must be used, reduce the Ibrutinib dose. Patients taking concomitant strong or moderate CYP3A4 inhibitors should be monitored more closely for signs of Ibrutinib toxicity .
- Avoid grapefruit and Seville oranges during Ibrutinib treatment, as these contain moderate inhibitors of CYP3A .
CYP3A Inducers
- Administration of Ibrutinib with rifampin, a strong CYP3A inducer, decreased ibrutinib Cmax and AUC by approximately 13- and 10-fold, respectively.
- Avoid concomitant use of strong CYP3A inducers (e.g., carbamazepine, rifampin, phenytoin and St. John's Wort). Consider alternative agents with less CYP3A induction
Use in Specific Populations
Pregnancy
Risk Summary
- Based on findings in animals, Ibrutinib can cause fetal harm when administered to a pregnant woman. If Ibrutinib is used during pregnancy or if the patient becomes pregnant while taking Ibrutinib, the patient should be apprised of the potential hazard to the fetus.
Animal Data
- Ibrutinib was administered orally to pregnant rats during the period of organogenesis at oral doses of 10, 40 and 80 mg/kg/day. Ibrutinib at a dose of 80 mg/kg/day was associated with visceral malformations (heart and major vessels) and increased post-implantation loss. The dose of 80 mg/kg/day in animals is approximately 14 times the exposure (AUC) in patients with MCL and 20 times the exposure in patients with CLL or WM administered the dose of 560 mg daily and 420 mg daily, respectively. Ibrutinib at doses of 40 mg/kg/day or greater was associated with decreased fetal weights. The dose of 40 mg/kg/day in animals is approximately 6 times the exposure (AUC) in patients with MCL administered the dose of 560 mg daily.
- There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Ibrutinib in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Ibrutinib during labor and delivery.
Nursing Mothers
- It is not known whether ibrutinib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Ibrutinib, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
- The safety and effectiveness of Ibrutinib in pediatric patients has not been established
.
Geriatic Use
- Of the 111 patients treated for MCL, 63% were 65 years of age or older. No overall differences in effectiveness were observed between these patients and younger patients. Cardiac adverse events (atrial fibrillation and hypertension), infections (pneumonia and cellulitis) and gastrointestinal events (diarrhea and dehydration) occurred more frequently among elderly patients.
- Of the 391 patients randomized in Study 2, 61% were ≥ 65 years of age. No overall differences in effectiveness were observed between age groups. Grade 3 or higher adverse events occurred more frequently among elderly patients treated with Ibrutinib (61% of patients age ≥ 65 versus 51% of younger patients) .
- Of the 63 patients treated for WM, 59% were 65 years of age or older. No overall differences in effectiveness were observed between these patients and younger patients. Cardiac adverse events (atrial fibrillation and hypertension), and infections (pneumonia and urinary tract infection) occurred more frequently among elderly patients.
Gender
There is no FDA guidance on the use of Ibrutinib with respect to specific gender populations.
Race
There is no FDA guidance on the use of Ibrutinib with respect to specific racial populations.
Renal Impairment
- Less than 1% of ibrutinib is excreted renally. Ibrutinib exposure is not altered in patients with Creatinine clearance (CLcr) > 25 mL/min. There are no data in patients with severe renal impairment (CLcr < 25 mL/min) or patients on dialysis
Hepatic Impairment
- Ibrutinib is metabolized in the liver. In a hepatic impairment study, data showed an increase in ibrutinib exposure. Following single dose administration, the AUC of ibrutinib increased 2.7-, 8.2- and 9.8-fold in subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B), and severe (Child-Pugh class C) hepatic impairment compared to subjects with normal liver function. The safety of Ibrutinib has not been evaluated in patients with hepatic impairment.
- Monitor patients for signs of Ibrutinib toxicity and follow dose modification guidance as needed. It is not recommended to administer Ibrutinib to patients with moderate or severe hepatic impairment (Child-Pugh classes B and C)
Females of Reproductive Potential and Males
- Advise women to avoid becoming pregnant while taking Ibrutinib because Ibrutinib can cause fetal harm
Immunocompromised Patients
Plasmapheresis
- Management of hyperviscosity in patients with WM may include plasmapheresis before and during treatment with Ibrutinib. Modifications to Ibrutinib dosing are not required.
Administration and Monitoring
Administration
- Oral
Monitoring
There is limited information regarding Monitoring of Ibrutinib in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Ibrutinib in the drug label.
Overdosage
Acute Overdose
There is limited information regarding Chronic Overdose of Ibrutinib in the drug label.
Pharmacology
This image is provided by the National Library of Medicine.

Mechanism of Action
- Ibrutinib is a small-molecule inhibitor of BTK. Ibrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. BTK's role in signaling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion. Nonclinical studies show that ibrutinib inhibits malignant B-cell proliferation and survival in vivo as well as cell migration and substrate adhesion in vitro.
Structure
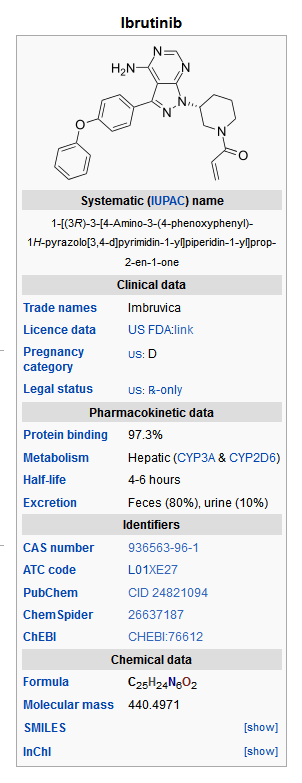
- Ibrutinib is an inhibitor of Bruton's tyrosine kinase (BTK). It is a white to off-white solid with the empirical formula C25H24N6O2 and a molecular weight 440.50. Ibrutinib is freely soluble in dimethyl sulfoxide, soluble in methanol and practically insoluble in water.
The chemical name for ibrutinib is 1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one and has the following structure:
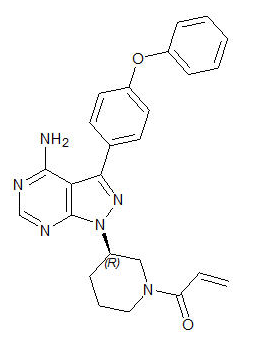
This image is provided by the National Library of Medicine.

- Ibrutinib capsules for oral administration are supplied as white opaque capsules that contain 140 mg ibrutinib as the active ingredient. Each capsule also contains the following inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate. The capsule shell contains gelatin, titanium dioxide and black ink. Each white opaque capsule is marked with "ibr 140 mg" in black ink.
Pharmacodynamics
- In patients with recurrent B-cell lymphoma > 90% occupancy of the BTK active site in peripheral blood mononuclear cells was observed up to 24 hours after ibrutinib doses of ≥ 2.5 mg/kg/day (≥ 175 mg/day for average weight of 70 kg).
Pharmacokinetics
Absorption
- Ibrutinib is absorbed after oral administration with a median Tmax of 1 to 2 hours. Ibrutinib exposure increases with doses up to 840 mg. The steady-state AUC (mean ± standard deviation) observed in patients at 560 mg is 953 ± 705 ng∙h/mL and in patients at 420 mg is 680 ± 517 ng∙h/mL. Administration with food increased ibrutinib Cmax and AUC by approximately 2 to 4- and 2-fold, respectively, compared with administration of ibrutinib after overnight fasting.
Distribution
- Reversible binding of ibrutinib to human plasma protein in vitro was 97.3% with no concentration dependence in the range of 50 to 1000 ng/mL. The volume of distribution at steady state (Vd,ss) was 683 L, and the apparent volume of distribution at steady state (Vd,ss/F) was approximately 10000 L.
Metabolism
- Metabolism is the main route of elimination for ibrutinib. It is metabolized to several metabolites primarily by cytochrome P450, CYP3A, and to a minor extent by CYP2D6. The active metabolite, PCI-45227, is a dihydrodiol metabolite with inhibitory activity towards BTK approximately 15 times lower than that of ibrutinib. The range of the mean metabolite to parent ratio for PCI-45227 at steady-state is 1 to 2.8.
Elimination
- Intravenous clearance was 62 and 76 L/h in fasted and fed conditions, respectively. In line with the high first-pass effect, the apparent oral clearance is approximately 2000 and 1000 L/h in fasted and fed conditions, respectively. The half-life of ibrutinib is 4 to 6 hours.
- Ibrutinib, mainly in the form of metabolites, is eliminated primarily via feces. After a single oral administration of radiolabeled [14C]-ibrutinib in healthy subjects, approximately 90% of radioactivity was excreted within 168 hours, with the majority (80%) excreted in the feces and less than 10% accounted for in urine. Unchanged ibrutinib accounted for approximately 1% of the radiolabeled excretion product in feces and none in urine, with the remainder of the dose being metabolites.
Age
- Age (37 to 84 years) does not alter ibrutinib systemic clearance.
Gender
- Gender does not alter ibrutinib systemic clearance.
Renal Impairment
- Ibrutinib is not significantly cleared renally; urinary excretion of metabolites is < 10% of the dose. Creatinine clearance > 25 mL/min had no influence on the exposure to Ibrutinib. There are no data in patients with severe renal impairment (CLcr < 25 mL/min) or in patients on dialysis.
Hepatic Impairment
- Ibrutinib is metabolized in the liver. In a hepatic impairment trial, a single dose of 140 mg of Ibrutinib was administered in non-cancer subjects. Ibrutinib AUC increased 2.7-, 8.2- and 9.8-fold, respectively, in subjects with mild (n=6), moderate (n=10) and severe (n=8) hepatic impairment relative to subjects with normal liver function. Ibrutinib Cmax increased 5.2-, 8.8- and 7.0-fold, respectively, in subjects with mild, moderate and severe hepatic impairment relative to subjects with normal liver function .
Drug Interactions
Coadministration of Ibrutinib with CYP3A Inhibitors
- In a sequential design trial of 18 healthy, fasted volunteers, a single dose of 120 mg of Ibrutinib was administered alone on Day 1 and a single dose of 40 mg of Ibrutinib was administered on Day 7 in combination with 400 mg of ketoconazole (given daily on Days 4 – 9). Ketoconazole increased ibrutinib dose-normalized Cmax and AUC 29-fold and 24-fold, respectively. Simulations using fasted conditions indicate that moderate CYP3A inhibitors diltiazem and erythromycin may increase AUC of ibrutinib by 5- to 8-fold.
Coadministration of Ibrutinib with CYP3A Inducers
- PK data from a dedicated drug interaction trial showed that rifampin (a strong CYP3A inducer) decreases ibrutinib Cmax and AUC by more than 13- and 10-fold. Simulations using PBPK suggested that a moderate CYP3A inducer (efavirenz) may decrease the AUC of ibrutinib by up to 3-fold.
Coadministration of Ibrutinib with CYP Substrates
- In vitro studies indicated that ibrutinib (I/Ki < 0.07 using mean Cmax at 560 mg) and PCI-45227 (I/Ki < 0.03) are unlikely to be inhibitors of any major CYPs at clinical doses. Both ibrutinib and the PCI-45227 are weak inducers of CYP450 isoenzymes in vitro.
Coadministration of Ibrutinib with Substrates of Transporters
- In vitro studies indicated that ibrutinib is not a substrate of p-glycoprotein (P-gp). Systemic ibrutinib is unlikely to be an inhibitor of P-gp at clinical doses ([I]1/Ki < 0.1). However, it may have an effect on P-gp substrates in the GI tract due to higher local concentrations after an oral dose. Co-administration of oral narrow therapeutic index P-gp substrates (e.g., digoxin) with Ibrutinib may increase their blood concentration.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment of Fertility
- Carcinogenicity studies have not been conducted with ibrutinib.
- Ibrutinib was not mutagenic in a bacterial mutagenicity (Ames) assay, was not clastogenic in a chromosome aberration assay in mammalian (CHO) cells, nor was it clastogenic in an in vivo bone marrow micronucleus assay in mice at doses up to 2000 mg/kg.
- Fertility studies with ibrutinib have not been conducted in animals. In the general toxicology studies conducted in rats and dogs, orally administered ibrutinib did not result in adverse effects on reproductive organs.
Clinical Studies
Mantle Cell Lymphoma
- The safety and efficacy of Ibrutinib in patients with MCL who have received at least one prior therapy were evaluated in an open-label, multi-center, single-arm trial of 111 previously treated patients. The median age was 68 years (range, 40 to 84 years), 77% were male, and 92% were Caucasian. At baseline, 89% of patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 42 months, and median number of prior treatments was 3 (range, 1 to 5 treatments), including 11% with prior stem cell transplant. At baseline, 39% of subjects had at least one tumor ≥ 5 cm, 49% had bone marrow involvement, and 54% had extranodal involvement at screening.
- Ibrutinib was administered orally at 560 mg once daily until disease progression or unacceptable toxicity. Tumor response was assessed according to the revised International Working Group (IWG) for non-Hodgkin's lymphoma (NHL) criteria. The primary endpoint in this study was investigator-assessed overall response rate (ORR). Responses to Ibrutinib are shown in Table 9.
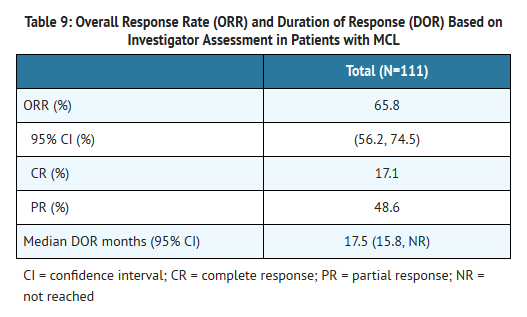
This image is provided by the National Library of Medicine.

- An Independent Review Committee (IRC) performed independent reading and interpretation of imaging scans. The IRC review demonstrated an ORR of 69%.
- The median time to response was 1.9 months.
Lymphocytosis
- Upon initiation of Ibrutinib, a temporary increase in lymphocyte counts (i.e., ≥ 50% increase from baseline and above absolute lymphocyte count of 5,000/mcL) occurred in 33% of patients in the MCL study. The onset of isolated lymphocytosis occurs during the first few weeks of Ibrutinib therapy and resolves by a median of 8 weeks.
Chronic Lymphocytic Leukemia
- The safety and efficacy of Ibrutinib in patients with CLL who have received at least one prior therapy were demonstrated in one uncontrolled trial and one randomized, controlled trial.
Study 1
- An open-label, multi-center trial was conducted in 48 previously treated CLL patients. The median age was 67 years (range, 37 to 82 years), 71% were male, and 94% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 80 months and the median number of prior treatments was 4 (range, 1 to 12 treatments). At baseline, 46% of subjects had at least one tumor ≥ 5 cm.
- Ibrutinib was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The ORR and DOR were assessed using a modified version of the International Workshop on CLL Criteria by an Independent Review Committee. The ORR was 58.3% (95% CI: 43.2%, 72.4%), all partial responses. None of the patients achieved a complete response. The DOR ranged from 5.6 to 24.2+ months. The median DOR was not reached.
Study 2
- A randomized, multicenter, open-label Phase 3 study of Ibrutinib versus ofatumumab was conducted in patients with previously treated CLL or SLL. Patients (n=391) were randomized 1:1 to receive either Ibrutinib 420 mg daily until disease progression, or unacceptable toxicity or ofatumumab at an initial dose of 300 mg, followed one week later by a dose of 2000 mg weekly for 7 doses and then every 4 weeks for 4 additional doses. Fifty seven patients randomized to ofatumumab crossed over following progression to receive Ibrutinib. The median age was 67 years (range, 30 to 88 years), 68% were male, and 90% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The trial enrolled 373 patients with CLL and 18 patients with SLL. The median time since diagnosis was 91 months and the median number of prior treatments was 2 (range, 1 to 13 treatments). At baseline, 58% of patients had at least one tumor ≥ 5 cm. Thirty-two percent of patients had 17p deletion.
- Progression free survival (PFS) as assessed by independent review committee (IRC) according to IWCLL criteria indicated a 78% statistically significant reduction in the risk of death or progression. Analysis of overall survival (OS) demonstrated a 57% statistically significant reduction in the risk of death for patients in the Ibrutinib arm. Efficacy results for Study 2 are shown in Table 10 and the Kaplan-Meier curves for PFS and OS are shown in Figures 1 and 2, respectively.
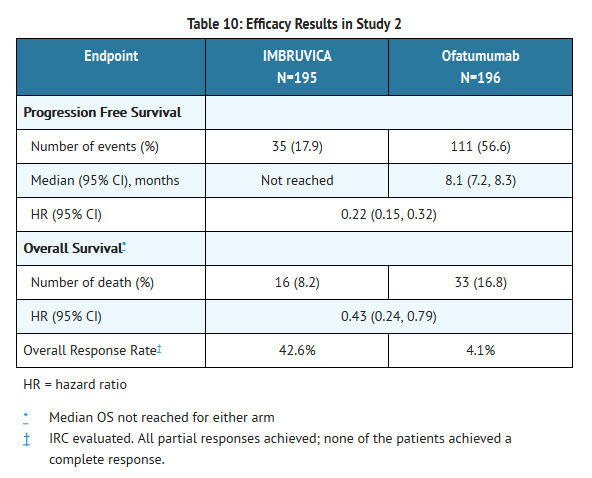
This image is provided by the National Library of Medicine. 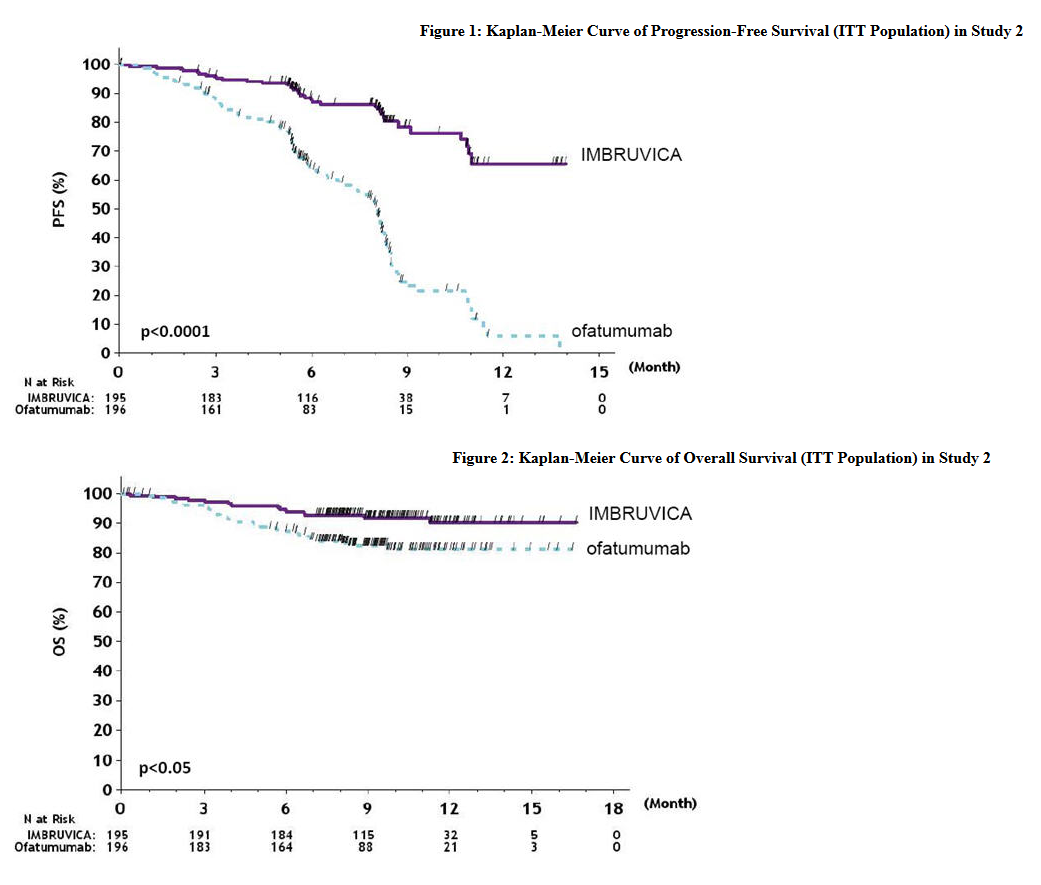
This image is provided by the National Library of Medicine.


CLL with 17p deletion (del 17p CLL)
- Study 2 included 127 patients with del 17p CLL. The median age was 67 years (range, 30 to 84 years), 62% were male, and 88% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. PFS and ORR were assessed by IRC. Efficacy results for del 17p CLL are shown in Table 11.
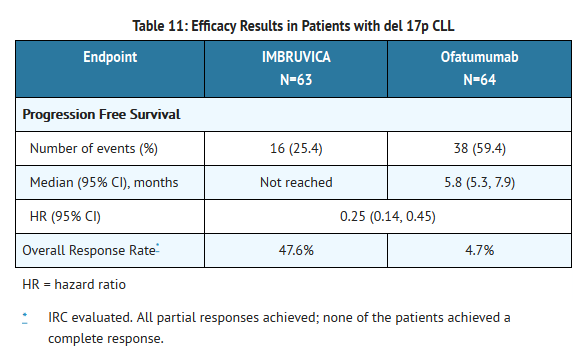
This image is provided by the National Library of Medicine.

Lymphocytosis
- Upon initiation of Ibrutinib, an increase in lymphocyte counts (i.e., ≥ 50% increase from baseline and above absolute lymphocyte count of 5,000/mcL) occurred in 77% of patients in the CLL study. The onset of isolated lymphocytosis occurs during the first month of Ibrutinib therapy and resolves by a median of 23 weeks (range 1 – 104+ weeks).
Waldenström's Macroglobulinemia
- The safety and efficacy of Ibrutinib in WM were evaluated in an open-label, multi-center, single-arm trial of 63 previously treated patients. The median age was 63 years (range, 44 to 86 years), 76% were male, and 95% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 74 months, and the median number of prior treatments was 2 (range, 1 to 11 treatments). At baseline, the median serum IgM value was 3.5 g/dL (range, 0.7 to 8.4 g/dL).
- Ibrutinib was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The responses were assessed by investigators and an Independent Review Committee (IRC) using criteria adopted from the International Workshop of Waldenström's Macroglobulinemia. Responses, defined as partial response or better, per IRC are shown in Table 12.
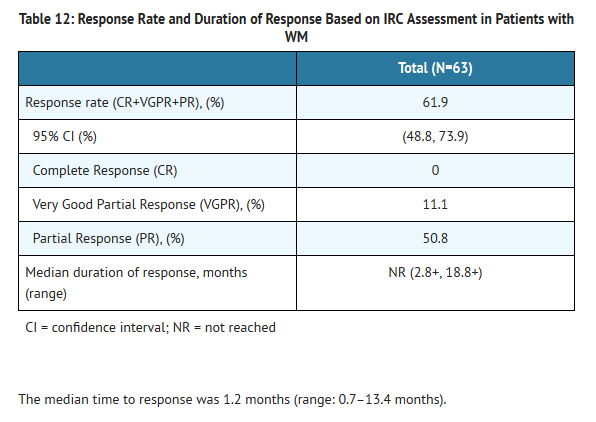
This image is provided by the National Library of Medicine.

How Supplied
- The white opaque 140 mg capsules marked with "ibr 140 mg" in black ink are available in white HDPE bottles with a child-resistant closure:
- 90 capsules per bottle: NDC 57962-140-09
- 120 capsules per bottle: NDC 57962-140-12
- Store bottles at room temperature 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C and 30°C (59°F to 86°F). Retain in original package until dispensing.
Storage
There is limited information regarding Ibrutinib Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Ibrutinib |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Ibrutinib |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
See FDA-approved patient labeling (PATIENT INFORMATION).
- Inform patients of the possibility of bleeding, and to report any signs or symptoms (blood in stool or urine, prolonged or uncontrolled bleeding). Inform the patient that Ibrutinib may need to be interrupted for medical or dental procedures .
- Infections:
- Counsel patients to report any signs of palpitations, lightheadedness, dizziness, fainting, shortness of breath, and chest discomfort .
- Second primary malignancies:
- Inform patients that other malignancies have occurred in patients who have been treated with Ibrutinib, including skin cancers and other carcinomas.
- Inform patients of the potential risk of tumor lysis syndrome and report any signs and symptoms associated with this event to their healthcare provider for evaluation
- Embryo-fetal toxicity:
- Advise women of the potential hazard to a fetus and to avoid becoming pregnant
- Inform patients to take Ibrutinib orally once daily according to their physician's instructions and that the capsules should be swallowed whole with a glass of water without being opened, broken, or chewed at approximately the same time each day.
- Advise patients that in the event of a missed daily dose of Ibrutinib, it should be taken as soon as possible on the same day with a return to the normal schedule the following day. Patients should not take extra capsules to make up the missed dose .
- Advise patients of the common side effects associated with Ibrutinib . Direct the patient to a complete list of adverse drug reactions in PATIENT INFORMATION.
- Advise patients to inform their health care providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products .
- Advise patients that they may experience loose stools or diarrhea, and should contact their doctor if their diarrhea persists. Advise patients to maintain adequate hydration.
Precautions with Alcohol
- Alcohol-Ibrutinib interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- Imbruvica®
Look-Alike Drug Names
There is limited information regarding Ibrutinib Look-Alike Drug Names in the drug label.
Drug Shortage Status
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Page Name=Ibrutinib
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Ibrutinib |Label Name=Ibrutinib12.png
}}
{{#subobject:
|Label Page=Ibrutinib |Label Name=Ibrutinib13.png
}}